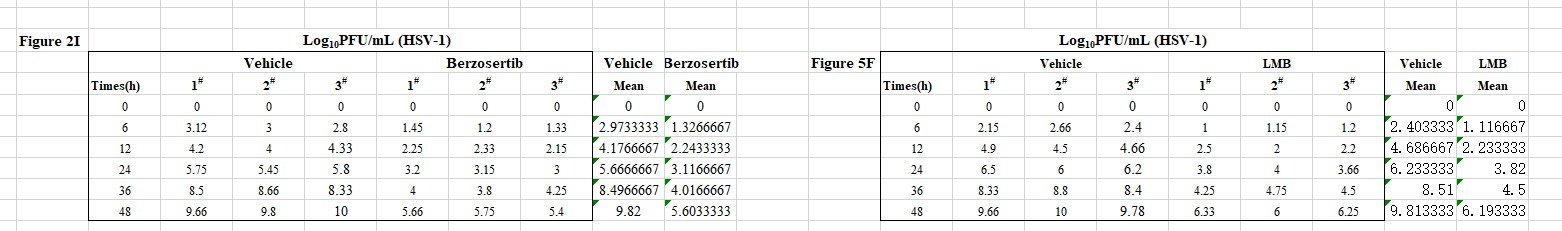
Author response:
Reviewer #1 (Public review):
Weaknesses:
While the breadth of techniques is impressive, the central premise of the work-the structural and functional relationship between polyQ assemblies and the Golgi apparatus-is not supported by sufficiently rigorous cell biological evidence.
A major concern is that much of the cell biology data remains descriptive and lacks mechanistic depth. The findings are fragmented and not integrated into a coherent molecular or cellular model. Instead of building a logical progression of experiments, the study presents a collection of observations that appear disconnected and, at times, driven more by technical capability than by hypothesis-driven design.
Critically, the key claim that polyQ HTT functionally disrupts the Golgi (Golgipathy) is not convincingly demonstrated. Many observations could be more simply explained by the polyQ HTT localization to the Golgi and known Golgi sensitivities to perturbations (e.g., starvation or Brefeldin A treatment), rather than by a specific mechanistic role of polyQ HTT.
The manuscript also suffers from issues in organization and clarity, including imprecise descriptions and figures that are difficult to interpret.
We thank the Reviewer for their time, valuable comments, and recognition of our technical expertise and resources. With our specialized background in pathology and super-resolution microscopy, our research heavily relies on structurally precise histological methods to address these fundamental biological questions. Furthermore, our laboratory maintains one of the largest repositories of patient-derived and healthy control fibroblasts, as well as iPSC lines, within the Huntington's disease (HD) research community. Because these patient-derived and engineered cell models express endogenous mutant HTT (mHTT) within an authentic genetic background, they provide a uniquely powerful system for decoding HD pathogenesis.
We appreciate the Reviewer’s comment regarding hypothesis-driven design. Classically, a hypothesis-driven approach relies on well-established, highly stable experimental platforms. However, a key finding of our study is the highly fragile and volatile nature of polyQ assemblies, particularly when subjected to post-fixation and oxidative stress. Because these structures can behave unpredictably under stress, we utilized an unbiased, data-driven approach leveraging our high-resolution imaging pipeline to explore polyQ assemblies in both healthy and HD cells.
Importantly, hypothesis-driven and data-driven methods are complementary rather than mutually exclusive. For instance, if a real-time tracking method were developed to endogenously label native HTT in living cells, it would open the door for direct hypothesis testing regarding polyQ assembly mechanics. Despite the current technical limitations of the field, our study successfully overcomes these challenges to reveal the spatial tomography and unique dynamics of polyQ assemblies directly within patient-derived cells. We fully discussed the limitations of this research in the discussion section.
We appreciate the Reviewer’s critical assessment regarding the functional disruption of the Golgi apparatus (Golgipathy). To rigorously investigate this phenomenon, we employed a comprehensive suite of methodologies ranging from live-cell imaging to single-cell RNA sequencing. Our findings build directly upon a well-established body of literature. We previously demonstrated that mutant huntingtin (mHTT) disrupts Golgi function within the neural tubes of human cortical organoids (hCO) (Liu et al., 2024), aligning with broader neurodevelopmental defects observed in HD (Barnat et al., 2020). Furthermore, prior independent studies have confirmed that both HTT knockdown and the presence of mHTT impair Golgi-to-plasma membrane trafficking, notably in primary fibroblasts from homozygous Htt<sup>140Q/140Q</sup> knock-in mice (Brandstaetter et al., 2014); mHTT also affects post-Golgi trafficking of proteins (del Toro et al., 2006). Backed by this literary consensus and our own multi-modal data, which Reviewer 2 also noted as sufficient, we are confident that our manuscript provides a robust, multi-layered demonstration of mHTT-induced Golgipathy.
In this study, we found that polyQ assemblies and the Golgi form a Golgi-polyQ complex mediated by ARF1 and ARFIP2. Thus, the structural coupling of polyQ assemblies with the Golgi apparatus under starvation, during the cell cycle, is rational.
Based on the reviewer suggestion, we will completely revise the entire manuscript. Hopefully, this revision will meet the requirement of smoothness and clarity.
Major Concerns:
(1) Golgi localization
The localization of polyQ HTT relies entirely on the antibody 3B5H10, which is foundational to the study. However, previous reports using the same antibody have described predominantly cytosolic localization. This discrepancy must be addressed rigorously by independent validation using alternative antibodies or tagged, exogenously expressed polyQ HTT constructs that should be shown to colocalize with 3B5H10 signals.
Despite historical inconsistencies across existing publications (Barnat et al., 2020; Hickman et al., 2022; Shen et al., 2019; Tousley et al., 2019), we noticed that the immunostaining results of multiple HTT antibodies are consistent with our data (DiFiglia et al., 1995; Ko et al., 2001; Velier et al., 1998; Wheeler et al., 2000). Although these pioneering studies lacked modern 3D high-resolution imaging and standardized staining protocols, their reported 2D distribution patterns heavily resemble our results. For instance, transmission electron microscopy (TEM) immunolabeling originally revealed that HTT localizes along Golgi cisternae (DiFiglia et al., 1995) and formed organized and parallel fibrils (DiFiglia et al., 1997). Furthermore, immunostaining with a panel of distinct antibodies, including MV2, 3, 4, 5, 6, and 1F8, demonstrated characteristic Golgi-like distribution patterns for HTT (Ko et al., 2001). In addition, our polyQ antibody immunostaining in human fetal brain, which is reflective of polyQ assembly, is nearly identical to the staining results of Barnat et al., publication in Science (Barnat et al., 2020).
We have carefully checked two early publications, which reported that 3B5H10 only binds expanded polyQ but does not bind a normal polyQ (non-disease causing), which displays a part of a neuron that has a cytosolic diffuse pattern of HTT in 3B5H10 staining (Legleiter et al., 2009; Miller et al., 2011). Based on our extensive experience with HTT immunohistochemistry, we hypothesize that this diffuse signal may reflect nonspecific background artifacts, often caused by high antibody concentrations, poor tissue fixation, inadequate post-incubation washing, or the presence of effete cells, or premature fragmentation of the polyQ tract prior to staining. Interestingly, Miller et al. utilized a rapid tissue-perfusion and sectioning protocol originally published in Brain Research Bulletin (Ko et al., 2001), which is optimized to preserve intact polyQ assemblies. When reviewing the original Brain Research Bulletin study (Ko et al., 2001), we noted that the immunostaining profiles for polyQ-containing HTT peptides (specifically using antibodies MW2, MW3, MW4, MW5, and 1F8) are entirely consistent with our data, yet completely diverge from the patterns reported by the Muchowski group (Legleiter et al., 2009; Miller et al., 2011) (please see Ko et al., 2001.Legleiter et al., 2009; Miller et al., 2011). Furthermore, contrary to the Muchowski group's claims, subsequent biophysical evidence by Owens et al. (2015) independently confirmed that 3B5H10 binds to both normal and expanded polyQ sequences in huntingtin exon 1 fusion proteins (Owens et al., 2015). Together, these observations strongly support the validity of our staining profiles.
Several antibodies, including MV1, 1C2, and 3B5H10, were previously reported to recognize the expanded, pathogenic polyQ tracts of HTT (Khoshnan et al., 2002; Miller et al., 2011; Wang et al., 2008). However, emerging studies reveal that these antibodies actually bind both short and long polyQ sequences (Klein et al., 2013; Owens et al., 2015). Because a standard antibody Fab epitode typically spans only 5 to 15 amino acids (or 3 to 4 sugar residues), and normal HTT polyQ repeats range from 18 to 24, it is theoretically impossible for an antibody to exclusively target expanded polyQ while sparing normal polyQ.
We previously noticed that 3B5H10 antibody immunostaining signals are located in the long projection of striatal neurons. As we did not notice an intact neuron in the two publications, we have no idea about the 3B5H10 antibody signals in the neuronal projections of those images.
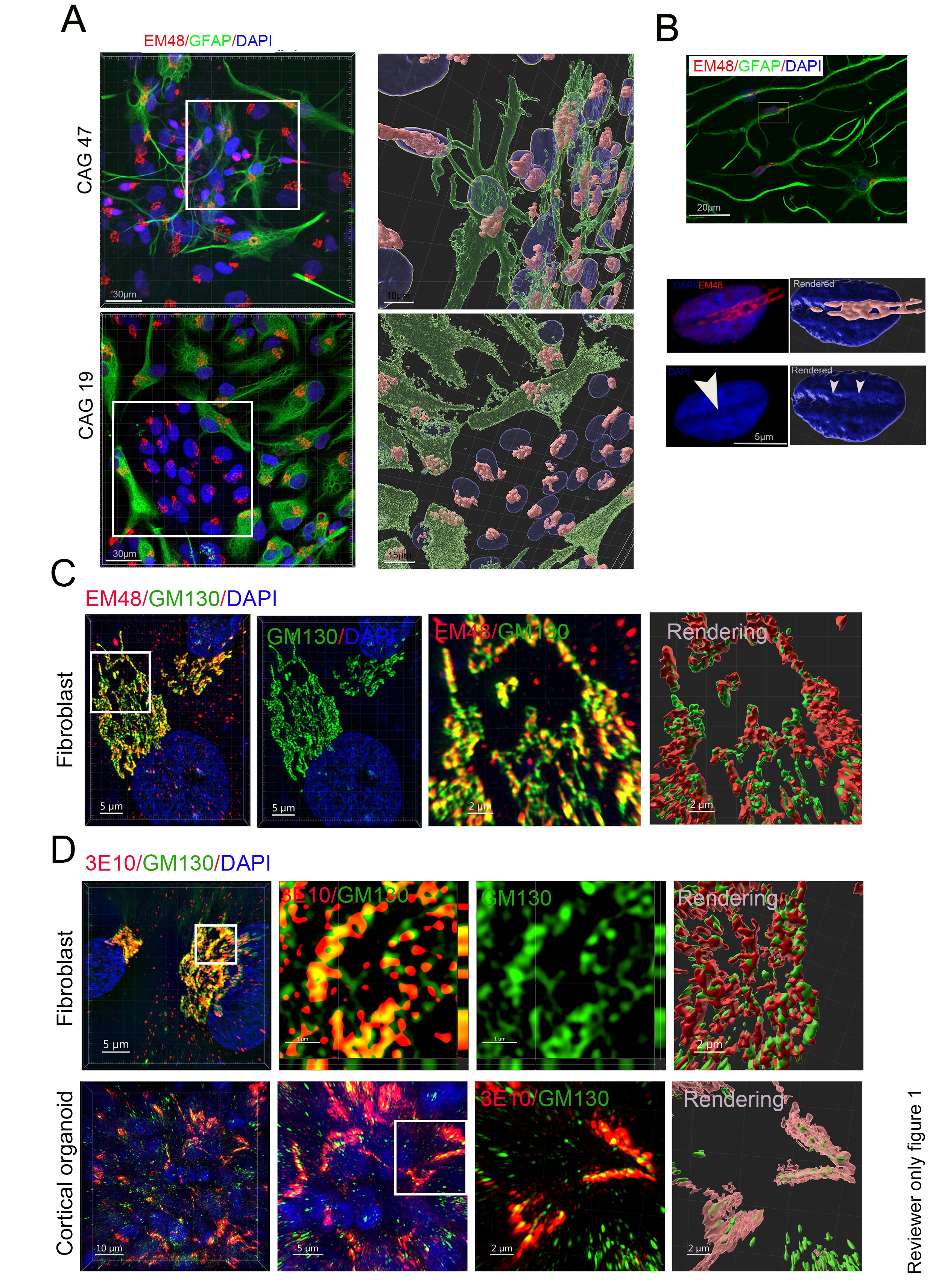
We investigated whether the polyQ assemblies detected by the 3B5H10 antibody contain full-length or large fragments of huntingtin (HTT). To test this, we selected two distinct HTT antibodies: EM48, which binds the first 256 amino acids (excluding the polyQ stretch), and 3E10, which targets the HDA region (amino acids 1171–1177). In patient fibroblasts, the immunostaining patterns for both EM48 and 3E10 were nearly identical to those observed with 3B5H10. These results demonstrate that the polyQ assemblies in fibroblasts are primarily composed of HTT proteins (see Author response image 1). We will include the results of EM48 and 3E10 immunostaining in the revised version.
Author response image 1.
(A) GFAP and EM48 antibodies staining of the astrocytes derived from HD patient and healthy sibling iPSCs showed polyQ assembly in the astrocytes derived from iPSC. (B). The spindle of polyQ assembly formed a dent on the nuclear surface of astrocytes. (C, D) Coimmunostaining of GM130 antibody with 3E10 or EM48 antibody in fibrobalsts revealed that polyQ assemblies contain great amount of HTTs. The middle and right panel are the sectional view boxed region (D) and the boxed region are a magnified part or rendering part (C, D) .
We also check whether the transfected exogenous HTT fragment of the first exon can be recruited into polyQ assemblies. We transfected fibroblasts with three vectors of the HTT first exon containing 19, 23, and 74 CAGs, respectively. The exogenous HTT fragments of the first exon did not significantly recruit into endogenous polyQ assemblies-Golgi complexes of fibroblasts (please refer to Reviewer only figure 5). We will include this part in the revised version.
In the cover letter, we told the editor that we have been studying this structure for over ten years. The results have been stable for over ten years.
Furthermore, the Golgi is identified solely using GM130, a cis-Golgi and ER exit site marker. This raises ambiguity: does polyQ HTT associate with the entire Golgi or only recruit GM130? Could the observed signal correspond to a sub-Golgi compartment?
Thank you for highlighting the precise sub-Golgi localization of GM130 as resolved by electron microscopy. We agree that transmission electron microscopy (TEM) demonstrates GM130 is restricted to the cis-Golgi network, intercisternal regions, and tubular structures, and is absent from the trans-Golgi (Nakamura et al., 1995). Given that individual Golgi cisternae measure approximately 20 nm in width, resolving cis- versus trans-Golgi sub-compartments exceeds the physical resolution limits of our microscopy system. Consequently, GM130 was utilized here as a robust, widely accepted pan-Golgi marker rather than a tool for sub-compartmental differentiation. To specifically evaluate the trans-Golgi network (TGN), we tracked Clathrin+ vesicles, which actively sort at the TGN (Klumperman, 2011). Our Clathrin staining confirms that polyQ assemblies localize to both the cis- and trans-Golgi compartments, as clearly demonstrated in the new lateral view projections provided in revised Figure 4C.
If polyQ HTT is indeed Golgi-associated, several key observations become expected rather than novel. For example, in Figure 4I-M, sensitivity to Brefeldin A is unsurprising, as Golgi structure collapses upon such treatment; in Figure 4N-O, co-fragmentation with the Golgi is expected under Golgi-disrupting conditions.
We agree that our data demonstrate the formation of a functionally coupled polyQ assembly–Golgi complex. Physically and structurally, the dynamics of polyQ assemblies are intrinsically linked to Golgi dynamics under distinct physiological states, including cell cycle progression and energy deprivation. This structural coupling is mediated by ADP-ribosylation factor 1 (ARF1). Specifically, the polyQ tract of HTT interacts with ARFIP2, which is one of the key effector proteins that physically bind active ARF1. Mechanistically, ARF1 is recruited to the Golgi membrane upon GDP-to-GTP exchange catalyzed by guanine nucleotide-exchange factors (GEFs). Consequently, treatment with Brefeldin A (BFA)—which inhibits ARF1 activation—effectively decouples the polyQ assemblies from both intact and fragmented Golgi structures.
Regarding the question of novelty, we define experimental novelty based on generating entirely unprecedented, empirical data that either confirms or redefines biological expectations, rather than evaluating conceptual expectations themselves. We believe the uncovering of this real-time, stimulus-responsive coupling mechanism provides fundamentally novel insights into HTT biology.
(2) 3D rendering
The extensive use of 3D rendering appears unnecessary and, in some cases, misleading. The rendered images do not provide additional insight beyond conventional 2D fluorescence images. Serial 2D fluorescence sections should be more objective in representing the 3D organization.
Thanks for pointing out the 3D rendering. While 3D rendering provides an essential spatial approximation of fluorescently labeled architectures, it offers significantly more precise structural information than conventional 2D or serial section imaging alone (Cao et al., 2023; Han et al., 2021; Hexige et al., 2015). A primary objective of our study was to evaluate these subcellular features within their intact, native three-dimensional context rather than relying solely on two-dimensional cross-sections. Crucially, without complete volumetric rendering, it is mathematically and visually challenging to accurately delineate complex morphological features, such as the nuclear gorge or true intranuclear accumulation. Consequently, 3D volumetric analysis and rendering are entirely indispensable for the accurate interpretation of the structural data presented in this study.
In Figure 2A and Figure 5A, red line features in 3D beige polyQ HTT structures resemble unrelated biological structures, such as vasculature, which is inappropriate.
We would like to clarify that there is no vasculature present within the referenced 3D rendering. The features the reviewer is highlighting are artifacts of the pseudo-coloring used exclusively to mask and visualize the surface tomography. In volumetric 3D rendering, pseudo-colors are assigned strictly to enhance visual clarity and contrast for the reader; they carry no intrinsic biological meaning or cellular identity. Furthermore, from a structural standpoint, the narrow red features in the rendered image are orders of magnitude smaller than true microvasculature. Functional microvessels possess a minimum diameter of 6 to 45 micrometers and exhibit a defined vascular lumen, endothelial cells, a basement membrane, pericytes, and a tunica intima. Therefore, based on both the scale of the image and established histological criteria, these features cannot biologically or structurally represent vasculature.
There is also an inconsistency in rendering. For example, fine mesh-like structures are shown in some figures (e.g., Figure 2A, Figure 4A), whereas others appear as amorphous aggregates (e.g., Figure 5A, Figure S2B), without explanation.
The selection of opacity and color masks in our 3D volumetric reconstructions is systematically chosen to optimize the visual clarity and spatial relationships between intersecting sub-cellular structures. For example, as shown in the fourth and fifth panels, an opaque blue mask was applied to clearly define the outer surface tomography. Conversely, in the third panel, a semi-transparent blue mask was utilized for the nucleus. This transparency is methodologically necessary because a subset of polyQ fragments is embedded within or localized directly inside the nuclear envelope; a transparent mask allows for the unambiguous visualization of these internal structures. Similarly, the inset in Figure 5A illustrates the distinct intranuclear occupancy pattern of polyQ, which also necessitates a transparent nuclear boundary. Collectively, these volumetric rendering strategies provide critical spatial and structural depth that cannot be captured by conventional 2D cross-sections or unconstructed serial imaging.
(3) Quantification of area and volume
The manuscript extensively quantifies the area and volume of polyQ assemblies (e.g., Figure 2B, C and Figure 3B, C, E, G, H). These measurements are not reliable. First, the structures appear filamentous and likely below the diffraction limit. Second, fluorescence signals are broadened by the point spread function (PSF), artificially inflating measured dimensions. Last, even with 3D SIM (~100 nm resolution), fine structural details remain unresolved. Thus, these quantitative measurements lack physical meaning and might not be used to support conclusions.
We appreciate the reviewer’s thoughtful critique regarding the quantification of area and volume. Our measurements are derived from immunofluorescent signals captured via structured illumination microscopy (SIM) and confocal imaging. If the reviewer's concern is that antibody-labeled structures do not perfectly match the absolute physical dimensions of native polyQ assemblies due to the linkage error of the primary-secondary antibody complex, we agree conceptually.
However, our imaging pipeline is optimized to minimize these discrepancies. Our SIM resolution reaches approximately 64 nm. Given that the total observed thickness of the fluorophore-labeled polyQ assemblies exceeds 200 nm, these structures reside well within the detectable range of our super-resolution system, minimizing diffraction-induced overestimation. Regarding the point spread function (PSF) and optical distortion, we emphasize that all comparative quantifications across experimental groups were conducted under identical imaging parameters and thresholds, ensuring a standardized baseline. Furthermore, our acquisition systems (Leica, Nikon, and Zeiss) utilize advanced deconvolution algorithms specifically designed to mitigate PSF-related blur. While we observed that deconvolution yielded negligible baseline improvements when using high-numerical-aperture objectives (63x) or 100x, oil immersion), it validates that our raw high-resolution scanning was already highly optimized.
We acknowledge that an immunolabeled complex is not structurally identical to a naked, pure polyQ tract. Nonetheless, indirect immunofluorescence remains the most robust method to evaluate spatial distribution in situ. Indeed, cryo-EM studies have highlighted that native polyQ tracts are highly flexible and structurally dynamic, making them exceptionally difficult to resolve in their native state (Guo et al., 2018). Intriguingly, we observed that antibody-bound polyQ assemblies remain structurally stable for several weeks with minimal fragmentation, suggesting that antibody binding may structurally stabilize these highly flexible regions. Consequently, indirect immunolabeling provides an indispensable framework for capturing these assemblies within the cellular environment.
(4) Interpretation of structural features (Figure 2A)
Descriptions such as "parallel spindles" and "ring-like assemblies" are not clearly supported by the data. The terminology is ambiguous, and the claimed structures are not discernible. The use of the term "interaction" with the nuclear membrane is also inappropriate. At best, the data suggest colocalization, which itself is not convincingly demonstrated.
Please refer to Fig. 2A (middle upper), Fig. 2F, and Fig. 4C for “parallel spindles”. Please refer to Fig. 5I, J, and Fig.S3C (right panel) for additional clear “ring-like assemblies”. Due to the unique spatial distribution of the 'ring-like assemblies', observing multiple rings within a single spindle is technically challenging. Accordingly, we have tempered our statement in the revised manuscript to accurately reflect this limitation. Furthermore, it is important to note that the visualized structures represent the fluorescent signal from secondary antibodies rather than direct imaging of the proteins themselves. Consequently, we cannot definitively confirm whether this immunostaining pattern precisely replicates the native state of polyQ assemblies within the cellular environment.
(5) Mitotic fragmentation (Figure 2E)
The conclusion that polyQ assemblies fragment during mitosis lacks proper controls. It is unclear whether these cells exhibited intact "fabric-like" assemblies during interphase, or the observed structures were already fragmented prior to mitosis.
We thought that we had displayed enough non-mitotic cells in this study (Fig. 2A, D, Fig. 4A, F). Most of the cells in this study are non-mitotic cells (G1+S+G2). Thus, we consider the control of non-mitotic cells to be redundant here.
(6) Fixation-induced fragmentation (Figure 2F)
The claim that fixation-induced fragmentation reflects a unique dynamic property of polyQ assemblies is likely an overinterpretation. This phenomenon may simply represent a fixation artifact. Therefore, it cannot be used as evidence for in-cellulo structural dynamics.
By definition, a laboratory artifact refers to any unintended structural detail, distortion, or error introduced by experimental equipment or the preparation process. We contend that the observed phenomenon represents a native chemical characteristic of the HTT polyQ domain inside cells following paraformaldehyde (PFA) fixation, rather than a technical artifact. Similar structural features have been documented by other investigators in tissue samples (Ferrante et al., 1997). A classic textbook example of an artifact is the lamina lucida of the basal lamina, which is artificially generated during electron microscopy tissue processing and does not exist in living tissue. In contrast, the fragmentation of polyQ assemblies occurs naturally both in living cells subjected to stress and during post-fixation processing.
(7) Nuclear localization claims (Figure 5A)
The assertion that polyQ assemblies "almost completely occupy the nucleus" is not supported. The images are more consistent with perinuclear localization, typical of the Golgi region. There is no clear evidence for nucleoplasmic distribution.
Please refer to the rendering image in the upper left inner insert of HD neurons (the blue [transparent] is the nucleus and the pink white is polyQ). The almost complete occupation of the nucleus is crystal clear in these images (rendered inner inserts, upper left). In iPSC-induced HD neurons, it is not only distributed in the nucleus but also in the cytoplasm. Based on your description, you might refer to the cytoplasmic polyQ assemblies but not the nucleus in the rendering image of the upper left (left panel). We will add a label in the revised version for clarity (white arrows for nuclear accumulation). In this manuscript, we have enough figures that clearly show the nuclear accumulation. Please also refer to Fig. 7 and Fig. S2 for additional images of nuclear accumulation.
(8) Drug treatment and data interpretation (Figure 3D-E)
The x-axis in Figure 3E is non-linear, which is inappropriate unless explicitly justified. Furthermore, the rationale for using Onjisaponin F is unclear. What is its known mechanism? Does it affect the Golgi organization? Without this context, observed effects may reflect Golgi perturbation rather than specific effects on polyQ assemblies.
We appreciate the reviewer pointing out Figure 3E. In this experiment, Huntington's disease (HD) fibroblasts were cultured in a low-glucose medium for the first 72 hours, which accounts for the linear trend observed across the first four data points. Following this 72-hour period, the cells were switched to a high-glucose medium and cultured for an additional 48 hours to evaluate subsequent dynamic changes in the polyQ assemblies. To improve visual clarity, we have color-coded these distinct treatment conditions in the revised manuscript, using red to denote low-glucose treatment and green to denote high-glucose treatment.
Regarding the choice of Onjisaponin treatment (a concern also raised by another reviewer), Onjisaponin is an active component derived from Radix Polygalae (Yuan Zhi). Previous literature indicates that Onjisaponin B enhances autophagy, accelerates the degradation of mutant α-synuclein and huntingtin in vitro, and activates the AMPK-mTOR signaling pathway (Wu et al., 2013). To optimize our experimental model, we screened multiple variants—specifically Onjisaponin B, D, and F. We determined that Onjisaponin F exhibits remarkably low cytotoxicity while maintaining a robust autophagy-enhancing capacity in both human fibroblasts and iPSC-derived neurons. Consequently, Onjisaponin F was selected for our human cell line experiments (please refer to the Reviewer only image 3). While we did not previously assess Golgi apparatus alterations under Onjisaponin F treatment, we recognize the value of this metric. We are currently evaluating changes to both the Golgi apparatus and neuronal firing rates following Onjisaponin F exposure, and this new dataset will be integrated into our revision.
Reviewer #2 (Public review):
[…] Overall, this work reports a novel polyQ assembly, which was previously reported as a pathogenic factor, has not been reported before for HTT, is related to Golgi activities and vesicular transport, and is dismantled in HD patient cells. The intensive immunostaining and super-resolution scanning are impressive and definitely strengthened by the impact of the findings. The scRNAseq data adds another layer to the observed Golgi impairments and their suggested relationship to Golgi function. The drug testing for polyQ assemblies, especially polyQ assemblies in HD cells, is preliminary. However, the data in this study are enough to support the existence of polyQ assemblies in human cells and their specific relationships with the Golgi apparatus.
We sincerely thank the reviewer for their time, dedication, and insightful evaluation of our manuscript. We agree that the drug screening component represents an initial phase of discovery, and we appreciate the opportunity to clarify this in our text. As the reviewer notes, executing high-throughput or exhaustive drug screenings in human brain organoids is exceptionally resource- and time-intensive due to prolonged culture requirements. We will provide more mechanistic and physiological details of these drug in the future.
Strengths:
In this study, the authors used the cells from a large HD family and fetal/child brain samples to decode the structure of endogenous polyQ assemblies. This part is impressive. The intensive staining and super-resolution scanning are amazing. The spatial relationships of polyQ assemblies with the Golgi apparatus and mitochondria are well illustrated.
Weaknesses:
Although they used healthy sibling cells as a control, an isogenic control (genetic correction of the mutant gene) is lacking. Based on the Golgipathy of mHTT, they did a drug screening. The drug testing for polyQ assemblies is preliminary. More rigorous validation, such as scRNA seq and proteomic analysis, etc., is necessary to reach a systemic conclusion.
References
Barnat, M., Capizzi, M., Aparicio, E., Boluda, S., Wennagel, D., Kacher, R., Kassem, R., Lenoir, S., Agasse, F., Braz, B.Y., et al. (2020). Huntington's disease alters human neurodevelopment. Science 369, 787-793.
Brandstaetter, H., Kruppa, A.J., and Buss, F. (2014). Huntingtin is required for ER-to-Golgi transport and for secretory vesicle fusion at the plasma membrane. Dis Model Mech 7, 1335-1340.
Cao, L., Ma, L., Zhao, J., Wang, X., Fang, X., Li, W., Qi, Y., Tang, Y., Liu, J., Peng, S., et al. (2023). An unexpected role of neutrophils in clearing apoptotic hepatocytes in vivo. Elife 12.
del Toro, D., Canals, J.M., Gines, S., Kojima, M., Egea, G., and Alberch, J. (2006). Mutant huntingtin impairs the post-Golgi trafficking of brain-derived neurotrophic factor but not its Val66Met polymorphism. J Neurosci 26, 12748-12757.
DiFiglia, M., Sapp, E., Chase, K., Schwarz, C., Meloni, A., Young, C., Martin, E., Vonsattel, J.P., Carraway, R., Reeves, S.A., et al. (1995). Huntingtin is a cytoplasmic protein associated with vesicles in human and rat brain neurons. Neuron 14, 1075-1081.
DiFiglia, M., Sapp, E., Chase, K.O., Davies, S.W., Bates, G.P., Vonsattel, J.P., and Aronin, N. (1997). Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 277, 1990-1993.
Ferrante, R.J., Gutekunst, C.A., Persichetti, F., McNeil, S.M., Kowall, N.W., Gusella, J.F., MacDonald, M.E., Beal, M.F., and Hersch, S.M. (1997). Heterogeneous topographic and cellular distribution of huntingtin expression in the normal human neostriatum. J Neurosci 17, 3052-3063.
Guo, Q., Bin, H., Cheng, J., Seefelder, M., Engler, T., Pfeifer, G., Oeckl, P., Otto, M., Moser, F., Maurer, M., et al. (2018). The cryo-electron microscopy structure of huntingtin. Nature 555, 117-120.
Han, X., Ma, L., Gu, J., Wang, D., Li, J., Lou, W., Saiyin, H., and Fu, D. (2021). Basal microvilli define the metabolic capacity and lethal phenotype of pancreatic cancer. J Pathol 253, 304-314.
Hexige, S., Ardito-Abraham, C.M., Wu, Y., Wei, Y., Fang, Y., Han, X., Li, J., Zhou, P., Yi, Q., Maitra, A., et al. (2015). Identification of novel vascular projections with cellular trafficking abilities on the microvasculature of pancreatic ductal adenocarcinoma. J Pathol 236, 142-154.
Hickman, R.A., Faust, P.L., Marder, K., Yamamoto, A., and Vonsattel, J.P. (2022). The distribution and density of Huntingtin inclusions across the Huntington disease neocortex: regional correlations with Huntingtin repeat expansion independent of pathologic grade. Acta Neuropathol Commun 10, 55.
Khoshnan, A., Ko, J., and Patterson, P.H. (2002). Effects of intracellular expression of anti-huntingtin antibodies of various specificities on mutant huntingtin aggregation and toxicity. Proc Natl Acad Sci U S A 99, 1002-1007.
Klein, F.A., Zeder-Lutz, G., Cousido-Siah, A., Mitschler, A., Katz, A., Eberling, P., Mandel, J.L., Podjarny, A., and Trottier, Y. (2013). Linear and extended: a common polyglutamine conformation recognized by the three antibodies MW1, 1C2 and 3B5H10. Hum Mol Genet 22, 4215-4223.
Klumperman, J. (2011). Architecture of the mammalian Golgi. Cold Spring Harb Perspect Biol 3.
Ko, J., Ou, S., and Patterson, P.H. (2001). New anti-huntingtin monoclonal antibodies: implications for huntingtin conformation and its binding proteins. Brain Res Bull 56, 319-329.
Legleiter, J., Lotz, G.P., Miller, J., Ko, J., Ng, C., Williams, G.L., Finkbeiner, S., Patterson, P.H., and Muchowski, P.J. (2009). Monoclonal antibodies recognize distinct conformational epitopes formed by polyglutamine in a mutant huntingtin fragment. J Biol Chem 284, 21647-21658.
Liu, Y., Chen, X., Ma, Y., Song, C., Ma, J., Chen, C., Su, J., Ma, L., and Saiyin, H. (2024). Endogenous mutant Huntingtin alters the corticogenesis via lowering Golgi recruiting ARF1 in cortical organoid. Mol Psychiatry.
Miller, J., Arrasate, M., Brooks, E., Libeu, C.P., Legleiter, J., Hatters, D., Curtis, J., Cheung, K., Krishnan, P., Mitra, S., et al. (2011). Identifying polyglutamine protein species in situ that best predict neurodegeneration. Nat Chem Biol 7, 925-934.
Nakamura, N., Rabouille, C., Watson, R., Nilsson, T., Hui, N., Slusarewicz, P., Kreis, T.E., and Warren, G. (1995). Characterization of a cis-Golgi matrix protein, GM130. J Cell Biol 131, 1715-1726.
Owens, G.E., New, D.M., West, A.P., and Bjorkman, P.J. (2015). Anti-PolyQ Antibodies Recognize a Short PolyQ Stretch in Both Normal and Mutant Huntingtin Exon 1. Journal of Molecular Biology 427, 2507-2519.
Paulson, H.L., Bonini, N.M., and Roth, K.A. (2000). Polyglutamine disease and neuronal cell death. Proc Natl Acad Sci U S A 97, 12957-12958.
Shen, M., Wang, F., Li, M., Sah, N., Stockton, M.E., Tidei, J.J., Gao, Y., Korabelnikov, T., Kannan, S., Vevea, J.D., et al. (2019). Reduced mitochondrial fusion and Huntingtin levels contribute to impaired dendritic maturation and behavioral deficits in Fmr1-mutant mice. Nat Neurosci 22, 386-400.
Tousley, A., Iuliano, M., Weisman, E., Sapp, E., Richardson, H., Vodicka, P., Alexander, J., Aronin, N., DiFiglia, M., and Kegel-Gleason, K.B. (2019). Huntingtin associates with the actin cytoskeleton and alpha-actinin isoforms to influence stimulus dependent morphology changes. PLoS One 14, e0212337.
Velier, J., Kim, M., Schwarz, C., Kim, T.W., Sapp, E., Chase, K., Aronin, N., and DiFiglia, M. (1998). Wild-type and mutant huntingtins function in vesicle trafficking in the secretory and endocytic pathways. Exp Neurol 152, 34-40.
Wang, C.E., Tydlacka, S., Orr, A.L., Yang, S.H., Graham, R.K., Hayden, M.R., Li, S., Chan, A.W., and Li, X.J. (2008). Accumulation of N-terminal mutant huntingtin in mouse and monkey models implicated as a pathogenic mechanism in Huntington's disease. Hum Mol Genet 17, 2738-2751.
Wheeler, V.C., White, J.K., Gutekunst, C.A., Vrbanac, V., Weaver, M., Li, X.J., Li, S.H., Yi, H., Vonsattel, J.P., Gusella, J.F., et al. (2000). Long glutamine tracts cause nuclear localization of a novel form of huntingtin in medium spiny striatal neurons in HdhQ92 and HdhQ111 knock-in mice. Hum Mol Genet 9, 503-513.
Wu, A.G., Wong, V.K., Xu, S.W., Chan, W.K., Ng, C.I., Liu, L., and Law, B.Y. (2013). Onjisaponin B derived from Radix Polygalae enhances autophagy and accelerates the degradation of mutant alpha-synuclein and huntingtin in PC-12 cells. Int J Mol Sci 14, 22618-22641.