While foresight will not reduce risk if no effectiveaction is available
Yes I fully agree and was saying this to my mother about 5 minutes ago before I got to this sentence
While foresight will not reduce risk if no effectiveaction is available
Yes I fully agree and was saying this to my mother about 5 minutes ago before I got to this sentence
There is a flash of value, followed by perpet-ual dusk or darkness
I believe this is what the world could head down if we suddenly invent AGI. The intelligence explosion would be huge, but we wouldn't have the means to be able to handle it. Reading groups like these are important to get the ball rolling to spark conversation about potential futures, but we must eventually (once we have enough context) be able to put in place safeguards and the infrastructure needed to move away from this path of a flash of value followed by perpetual darkness.
An example of thiskind is a scenario in which machine intelligence replacesbiological intelligence but the machines are constructedin such a way that they lack consciousness (in the senseof phenomenal experience)
This is what is happening now! The way these frontier AI models are built is fundamentally flawed if we are looking to achieve consciousness. I often like referencing Jeff Hawkins. We first must understand the brain before trying to replicate it. These LLMS as we most all know, are just intelligent autofill. Sure they can replace the manual labor of humans tenfold, but they will not and cannot invent the warpdrive.
it is about control
This reminds me of Dr. Parker's comment/question to Dr. Bialystok when she joined our class about whether AI is, in fact, a grand plan/conspiracy for control. Dr. Bialystok seemed to reject this idea, stating that she didn't think these young programmers were thinking that far ahead, but I wonder if the same can be said for university spaces. Even if AI isn't an overall conspiracy, perhaps the ways in which it is adopted in certain spaces are more plausible instances of control tactics.
more serious threat to university scholarship
yes, the scholarslop is a serious threat. I will argue, though, that AI can be used intelligently to advance knowledge and improve outputs, but not without discernment and critical thinking skills.
materials stripped of the specificity and disciplinary expertise that characterise the best academic teaching
This sentence made me think of how academics are treated with respect to academic integrity cases, at least at the University of Windsor. Instructors have no power at all. Though we are the ones with subject expertise and the ones designing and teaching classes and assessing student work, we are not allowed to decide on any sanctions for academic dishonesty. Rather, we are expected to report the alleged offense, through forms and evidence, which is then assessed by the Office of Graduate Studies (who are very much removed from the situated experience in the classroom involved). All of this is to say that institutions have already been treating academics this way in other spaces -- our knowledge and expertise are not trusted/respected, and decisions are made outside of situated spaces.
What matters here is not
I would argue that this absolutely does matter. If they demonstrated genuine understanding or intellectual insight, we'd be having a much different conversation (i.e., can we vs. should we). Using a flawed tool as truth is what is so dangerous here!
but the academic wielded disciplinary
AI still does not have this situated knowledge. It can come up with convincing arguments, but it is our choice whether to accept them or dig in and critique their veracity.
Varieties of Non-Standardized English spoken in some communities of color, like African American Vernacular English(which has also been called Ebonics) and Caribbean English Creole, have traditionally been seen asincorrect and inferior, especially by schools and government institutions.
Sometimes schools act as though standard English is the only right type of English, which causes students who speak other dialects to feel inferior.
declines are pretty consistent
Power structures survive on others not having power. Education is power. Knowledge is power. If we devalue post-secondary education, people have less power, voice, and agency to disrupt these dominant systems. It's somewhat unsurprising that this is "not a partisan thing."
That, and nothing else
This is interesting. I wonder, if the public believed in the value of post-secondary education, would it make a difference? Would governments change their tune? Until we can show people (governments, public) what's "in it for them," this attitude will continue. So, perhaps that is precisely what we should be focusing on with respect to external relations. It's tragic, though, that gov't/the public no longer view post-secondary education as a benefit to the greater good.
I'm also curious, though -- students are still enrolling in PSE -- why, if they don't see the value?
budget constraint
production possibility frontier or budget constraint.
all possible combinations of two goods you can purchase for a given budget
Globalization
reasons: faster / cheaper: - ship / air cargo - computing / information sharing - int. agreements
Most economies in the real world are mixed
three types of economies:
tradition - do things have have been done in the past
command - do things as instructed by a ruler
market - do things according to the needs and wants of the population
specialization
allows focus on the parts of production where they have an advantage
when people specialise in what they do best, they produce more than a combination of things
they learn to produce faster & better
business has economies of scale. i.e wrights law, or the learning rate of some industry
requires trade.
division oflabor
a good/service is broken into smaller steps and completed by different workers
modal verb
Expresiones de Certeza o Imposibilidad:
Pistas: "I am certain that...", "I am sure he didn't...", "It is impossible that...", "There is no doubt that..."
Significado: Exige modales de deducción (must have + participio, can't have + participio) o expresiones de certeza (be bound to).
Expresiones de Probabilidad o Expectativa:
Pistas: "It is probable that...", "People expect him to...", "It was arranged that..."
Significado: Exige estructuras con adjetivos modales (be likely to, be supposed to, be due to).
Expresiones de Acción Innecesaria en Pasado:
Pistas: "It was not necessary to do X, but they did it anyway..."
Significado: Exige la estructura needn't have + participio.
linking words and phrases
Presencia de conectores simples o informales:
Pistas: "although", "because", "if", "as soon as", "in order to", "so that".
Significado: La frase te exige elevar el nivel formal usando una locución avanzada o una estructura fija con preposición.
Estructuras de inmediatez o consecuencia directa:
Pistas: "Immediately after...", "The moment that...", "As soon as..."
Significado: Sugiere el uso de un conector de inversión (No sooner... than, Hardly... when).
Condiciones o restricciones:
Pistas: "Only if...", "On the condition that...", "If you don't..."
Significado: Sugiere conectores como provided (that), as long as, unless, o inversión condicional (Should you..., Were you to...).
cleft sentences
A. It-Clefts (Énfasis en un elemento específico)
Sirven para destacar quién hizo algo, cuándo ocurrió o qué elemento en concreto fue el afectado.
Estructura: It + is/was + [elemento enfatizado] + that / who + [resto de la oración]
Ejemplo original: Sarah designed the new logo.
Cleft: It was Sarah who designed the new logo.
B. Wh-Clefts / Pseudo-Clefts (Énfasis en la acción o el objeto)
Sirven para resaltar lo que alguien hizo, le gustó o necesitó.
Estructura: What + [Sujeto] + [Verbo] + is/was + [elemento enfatizado]
Ejemplo original: I really need a good cup of coffee.
Cleft: What I really need is a good cup of coffee.
C. All-Clefts (Énfasis en "lo único que...")
Sustituyen oraciones con only o just.
Estructura: All (that) + [Sujeto] + [Verbo] + is/was + [elemento enfatizado]
Ejemplo original: He only wants a second chance.
Cleft: All he wants is a second chance.
D. Clefts de Razón, Persona, Lugar o Tiempo
Sustituyen explicaciones largas usando sustantivos de categoría.
Razón: The reason why + [oración] + is/was + [que / sustantivo]
Tiempo / Momento: It was not until + [tiempo/evento] + that + [oración]
participle clauses
Conectores de causa: because, since, as.
Conectores de tiempo / secuencia: after, before, while, when, as soon as, once.
Conectores de condición: if, unless.
past tenses
-Expresiones de preferencia sobre acciones ajenas o pasadas: Significado: Requiere el uso de would rather / would sooner + Past Simple (si es presente/futuro) o Past Perfect (si es pasado).
Indicaciones de urgencia o retraso: Significado: Exige la estructura It's high time / It's about time + Past Simple.
Secuencia de acciones en el pasado con duración: Significado: Exige pasar del tiempo simple o continuo al Past Perfect Continuous (had been working).
fixed expressions
La palabra clave suele ser un sustantivo abstracto, un adjetivo o un verbo que "exige" una preposición o complemento rígido a su alrededor.
first we need to identify whatcontent from the 1° sentence has already been included in the 2" sentence.
step 1- Identifying similar content
The 1°* sentence and the 2"? sentence always use the same grammar.Your final answer will have the same meaning as the original sentence.The key word must be used in your answer.You can change the key word (e.g. to make it plural, to change the tense).An answer with five words is possible.
Understanding the instructions
So what makes a technically newer brand of typewriter more valuable than an older one?
reply to liamkembleyoung at https://reddit.com/r/typewriters/comments/1v4ykv9/pros_and_cons_of_pre1950s_typewriters_vs_1950s/
Quality of the materials, build, and engineering, condition of the machine (clean, well-adjusted, rubber condition), popularity, rarity, desirability, collectability all figure into machine pricing.
Most would say that the $250-350 Royal Classic/We R Memory Keepers machines from Shanghai Weilv Mechanism Company that are available new from Amazon and other retailers are cheap, poorly made pieces of junk with dreadful quality control and thus not worth the money one would spend on them. (The fact that they sell on ShopGoodWill for $5-10 underlines this.)
You might also have a 1970s Brother typewriter (made in the millions) that might have sold brand new in the $100 range versus a used 1955 Olympia SG1 selling in that same shop at the same time (1970s) for about the same money, or possibly less. The SG1 is acknowledged as one of the finest typewriters ever manufactured versus the very pedestrian and "cheaper" (materials-wise) Brother, so it's going to be worth more money now in the secondary market. The SG1 is less common, has a better reputation, more popular, more collectible, and just a better machine overall which is also likely to last longer. At the low end the SG1 is going to go for $75-150 while the Brother will be $25. Fully serviced out of a repair shop (cleaned, oiled, and adjusted with new rubber feet and a recovered platen) they might go respectively for $700-850 and $400-475.
Keep in mind that most typewriters you'll see on this sub and in the broader market are far from "rare"; rarity and desirability are two different things; and rare doesn't always equate to being more expensive. Actually rare machines aren't often discussed on this sub in general.
The signs by these once read ‘STOP WHILE LIGHTS FLASH’ but after a number of accidents the signs were changed in parts of northern England. Apparently in some areas the word while is used to mean until. Some drivers, therefore, had stopped until the lights flashed and then driven onto the railway line!
This is the beauty of different languages and dialects, it puts into example the hidden indifferences, although both dialects have the same words they have different meanings depending on where your from.
However, it would be foolish to ignore the fact that some dialects have greater status than others.
There is a silent conscious that there's a hierarchy of languages. it's never been a set rule but people make up their own tiers.
Twenty years
should read: The creative, production, and execution behind every StoryCycle engagement is powered by Bowstring's Twenty Five of storytelling, and production.
Peer reviewer use of AI is unethical because confi-dentiality of the manuscript cannot be maintained.When accepting the review invitation, reviewers areinstructed not to share the manuscript
Can still be leaked either way.
offer them an encouraging word and affirm somestrengths of their work.
Shows slight bias in the author as if AI gave peer review that showed weakness and where to improve the call for encouraging words would be nonexistent.
If we lose the writer, we lose the nurse”
The same idea can be translated into an educational setting.
When we let AI do that work for us, we’renot saving time; we’re outsourcing our thinking. Evenworse, we’re diluting the voice of nursing itself.”
In a way this can support my idea that if AI is used to assist writing to a certain point, it is truly still human words and work that end up being published?
but I will point out dangers of using AI: (1)to search scholarly literature, i2) to write manuscripts report-ing one’s own research or scholarly projects, and (3) to con-duct peer reviews of journal manuscript submissions.
Main idea that the text is based around.
Artificial intelligence has become a plague in higher educa-tion, with professors bemoaning AI-generated student essays,
This supports my earlier claim in the real world AI usage has no real way of being restricted besides running work through an AI detector which can still be fooled.
AI tools can only pro-duce information based on their training (which caninclude racial or gender bias)
This helps to further the claim that AI only knows what it has been taught by people, that is why bias and in many cases this inability to detect sham papers can exist
(one that exists to profit from large fees paidby authors, providing little or no peer review of arti-cle content)
Would additional guidelines to a prompt not prevent this?
I argue that Artificial Intelligence tools cannotbe trusted and should not be used for scholarly literaturesearches, for the following reasons:
This article directly disagrees with "AI Is Like 50% Good and 50% Bad": Exploring the Strengths and Challenges of AI in the Elementary Writing Classroom.” And its claim that AI can effectively be used in the classroom and in its specific case to review writings created by students.
worked" asks for a count of hours, not a clock time. From 8:00 AM to 5:00 PM is 9 hours. She takes a 1-hour meal break, so she writes 8. Worked example: a leave form
The text can be expanded beyond simple text/label matching. For instance, in Rosario’s time card example, explicitly demonstrating how to account for break deductions ( 9 total elapsed hours - 1 hour break = 8 working hours. This helps reinforce how word recognition and numerical reasoning interact on technical forms.
ere is how Rosario fills it in: She starts cooking at 8:00 in the morning. "Time in" means the start time, so 8:00 AM goes there.She leaves at 5:00 in the afternoon. "Time out" means the leaving time, so 5:00 PM goes there."Hours worked" asks for a count of hours, not a clock time. From 8:00 AM to 5:00 PM is 9 hours. She takes a 1-hour meal break, so she writes 8.
Again there is a need to use an english tone applicable to Filipino Learners, for ex:
1.She starts working at 8:00 in the morning. "Time In" refers to the exact time work starts, so she enters 8:00 AM.
She leaves work at 5:00 in the afternoon. "Time Out" refers to the time work ends, so she enters 5:00 PM.
This refers to the total number of work hours, not a specific clock time. From 8:00 AM to 5:00 PM is 9 hours. Subtracting her 1-hour lunch break, her total time worked is 8 hours.
StoryCycle is the operating system that fixes this. Not a campaign, not a rebrand. One unified business story, activated across every channel and team, measured against pipeline conversion.
should we stack this under the other paragraph?
Taylor,
https://bib.vincent-bonnefille.fr/search/stored?query=Charles+Taylor
« rend des avis dans l’intérêt de la loi et du bien commun ».
Article L432-1
Version en vigueur depuis le 20 novembre 2016
Modifié par LOI n°2016-1547 du 18 novembre 2016 - art. 40 Modifié par LOI n°2016-1547 du 18 novembre 2016 - art. 41
Le procureur général porte la parole aux audiences des chambres mixtes et de l'assemblée plénière ainsi que dans les assemblées générales de la cour.
Il peut la porter aux audiences des chambres et devant les formations prévues à l'article L. 441-2.
Il rend des avis dans l'intérêt de la loi et du bien commun. Il éclaire la cour sur la portée de la décision à intervenir.
fins qui sont d’abord individuelles ».
https://www.erudit.org/fr/revues/ps/1998-v17-n1-2-ps2491/040115ar.pdf
https://www.persee.fr/doc/assr_0335-5985_1998_num_102_1_1116_t1_0134_0000_4
« post-libéral ».
Ouverture
plete the pre-course survey. This page explains how to access the mobile app, how to download/save content an
this is good
ablished the stakes of
testtt
Chapters 1 a
testing tags
eLife Assessment
This manuscript reports an important new statistical method for calculating the significance of correlations between two time-series, which provides more accuracy than other methods when the data has few replicates. The proposed method solves a real-life problem that is frequently encountered and is broadly applicable to many realistic datasets in many experimental contexts. The technique is supported with compelling mathematical derivations as well as analysis of both computer-generated and previously published experimental data.
Reviewer #1 (Public review):
[Editors' note: this version has been assessed by the Reviewing Editor without further input from the original reviewers. The authors have addressed the comments raised in the previous round of review: Definitions and terminology have been made more precise. Additional analysis confirms the conclusions previously stated and clarifies concerns about the computational tractability of the method.]
Summary:
The manuscript puts forward a statistical method to more accurately report the significance of correlations within data. The motivation for this study is two-fold. First, the publication of biological studies demands the report of p-values, and it is widely accepted that p-values below the arbitrary threshold of 0.05 give the authors of such studies justification to draw conclusions about their data. Second, many biological studies are limited by the number of replicate samples that are feasible, with replicates of less than 5 typical. The authors report a statistical tool that uses a permute-match approach to calculate p-values. Notably, the proposed method reduces p-values from around 0.2 to 0.04 as compared to a standard permutation test with a small sample size. The approach is clearly explained, including detailed mathematical explanations and derivations. The advantage of the approach is also demonstrated through analysis of computer-generated synthetic data with specified correlation and analysis of previously published data related to fish schooling. The authors make a clear case that this method is an improvement over the more standard approach currently used and also demonstrate the impact of this methodology on the ability to obtain p-values that are the standard for biological research. Overall, this paper is very strong. While the subject matter seems somewhat specialized, I would make the case that this will be an important study that has broad general interest to readers. The findings are very general and applicable to many research contexts. Experimentalists also want to report accurate p-values in their work and better understand how these values are calculated. Although I believe the previous statement is true, I am not sure that many research groups doing biological work are reading specialized statistics journals regularly. Therefore, a useful and broadly applicable statistical tool is well placed in this journal.
Strengths:
The proposed method is broadly applicable to many realistic datasets in many experimental contexts.
The power of this method was demonstrated with both real experimental data and "synthetic" data. The advantages of the tool are clearly reported. The zebrafish data is a great example dataset.
The method solves a real-life problem that is frequently encountered by many experimental groups in the biological sciences.
The writing of the paper is surprisingly clear, given the technical nature of the subject matter. I would not at all consider myself a statistician or mathematician, but I found the text easy to follow. The authors did an impressive job guiding the reader through material that would often be difficult to grasp. The introduction was also well-written and clearly motivated the goals of the study.
Reviewer #2 (Public review):
Summary:
This paper presented a hypothesis testing procedure for the independence of two time-series that was potentially suitable for nonlinear dependence and for small-sample cases. This should bring potential benefits for biology data.
Strengths:
The test offers good flexibility for different kinds of dependence (through adjusting \rho) and seems to have good finite sample performance compared to the literature. The justification regarding the validity of the test procedure is clear.
Author response:
Public Reviews:
Reviewer #1 (Public review):
Summary:
The manuscript puts forward a statistical method to more accurately report the significance of correlations within data. The motivation for this study is two-fold. First, the publication of biological studies demands the report of p-values, and it is widely accepted that p-values below the arbitrary threshold of 0.05 give the authors of such studies justification to draw conclusions about their data. Second, many biological studies are limited by the number of replicate samples that are feasible, with replicates of less than 5 typical. The authors report a statistical tool that uses a permute-match approach to calculate p-values. Notably, the proposed method reduces p-values from around 0.2 to 0.04 as compared to a standard permutation test with a small sample size. The approach is clearly explained, including detailed mathematical explanations and derivations. The advantage of the approach is also demonstrated through analysis of computer-generated synthetic data with specified correlation and analysis of previously published data related to fish schooling. The authors make a clear case that this method is an improvement over the more standard approach currently used, and also demonstrate the impact of this methodology on the ability to obtain p-values that are the standard for biological research. Overall, this paper is very strong. While the subject matter seems somewhat specialized, I would make the case that this will be an important study that has broad general interest to readers. The findings are very general and applicable to many research contexts. Experimentalists also want to report accurate p-values in their work and better understand how these values are calculated. Although I believe the previous statement is true, I am not sure that many research groups doing biological work are reading specialized statistics journals regularly. Therefore a useful and broadly applicable statistical tool is well placed in this journal.
Strengths:
The proposed method is broadly applicable to many realistic datasets in many experimental contexts.
The power of this method was demonstrated with both real experimental data and "synthetic" data. The advantages of the tool are clearly reported. The zebrafish data is a great example dataset.
The method solves a real-life problem that is frequently encountered by many experimental groups in the biological sciences.
The writing of the paper is surprisingly clear, given the technical nature of the subject matter. I would not at all consider myself a statistician or mathematician, but I found the text easy to follow. The authors did an impressive job guiding the reader through material that would often be difficult to grasp. The introduction was also well-written and clearly motivated the goals of the study.
We appreciate the reviewer’s summary of our study and its strengths.
Weaknesses:
A few changes could be made if the manuscript is revised. I would consider all of these points minor, but the paper could be improved if these points were addressed.
(1) The caption of Figure 2 doesn't seem to mention panel D. Figure A-2 also does not mention C in the caption.
We apologize for this error, and thank you for catching it! The figure legends had missing or incorrect panel labels. This error has been corrected.
(2) Figure 2D is a little hard to follow. First, the definition of "Power" is not clear, and I couldn't find the precise definition in the text. Second, the legend for the different lines in 2D is only given in Figure A-2. Perhaps a portion of the caption for Figure 2 is missing?
We have added a definition of power in the main text:
“Although the permutation test, simultaneous permute-match test, and sequential permute-match test are all valid, they vary in power – the probability of detecting true dependence.”
We have clarified the use of “power” in legend of Fig 2 and clarified that the color key for Fig 2D is in Fig 2A. The relevant excerpt of the Fig 2 legend is copied here:
“(D) Statistical power for the permutation test and various permute-match tests as a function of the replicate number n, significance level α, and strength of dependence r<sub>X, Y</sub>. Power was estimated as the proportion of simulations in which dependence was detected, calculated from 5000 simulations at each value of r<sub>X, Y</sub> between r<sub>X, Y</sub> = 0 and 0.54 in steps of size 0.01. At r<sub>X, Y</sub> = 0, there is no dependence, so the curve at that point indicates the false positive rate rather than power. We chose the Pearson correlation coefficient as our correlation function ρ. See (A) for the color legend.”
We have also added dotted lines connecting the legend in panel A to the curves in panel D.
(3) The concept of circular variance for the fish data was heard to understand/visualize. The equation on line 326 did not help much. If there is a very simple picture that could be added near line 326 that helps to explain Ct and theta, that could be a big help for some readers who do not work on related systems. The analysis performed is understandable, the reader just has to accept that circular variance captions the degree of alignment of the fish.
We have replaced references to circular concentration with “mean resultant length”, which is the standard jargon for this term in circular statistics, and we have added an illustration.
(4) For the data discussed in Figure 3, I wasn’t 100% sure how the time windows were selected. In the caption, it says “time series to different lengths starting from the first frame”. So the 20 s time window was from t=0 to t= 20 s. Would a different result be obtained if a different 20 s window was chosen (from t = 4 min to t = 4 min 20 s just to give a specific example). I suppose by chance one of the time windows would give a pvalue less than the target 0.05, that wouldn’t be surprising. Maybe a random time window should be selected (although I am not indicating what was reported was incorrect)? A little more discussion on this aspect of the study may be helpful.
As suggested by the reviewer, we have redone the analysis of Figure 3D with random segments. This provides a more complete picture of how the chance of detecting a significant correlation varies with segment length. The main conclusion is unchanged: Perfect match tests reliably detect dependence across a wider range of segment lengths than the naive parametric alternative.
The relevant panel and an excerpt from the legend text are copied below.
“(D) Permute-match tests detected a significant correlation between speed and alignment more consistently than the parametric test. For a grid of lengths between 20 and 600 seconds we sampled 500 random segments of each length, each drawn from the first 600 seconds, and determined for each segment whether the parametric test and/or the two possible permute-match tests detected a significant (p ≤ 0.05) correlation. In the edge case of the maximum 600-second length, all 500 “random” segments were identical.”
Reviewer #2 (Public review):
Summary:
This paper presented a hypothesis testing procedure for the independence of two timeseries that was potentially suitable for nonlinear dependence and for small-sample cases. This should bring potential benefits for biology data.
Strengths:
The test offers good flexibility for different kinds of dependence (through adjusting \rho), and seems to have good finite sample performance compared to the literature. The justification regarding the validity of the test procedure is clear.
We appreciate the reviewer’s summary of key aspects of our manuscript.
Weaknesses:
(1) The size of the test is not guaranteed to (asymptotically) equal \alpha, which may damage the power.
We thank the reviewer for raising the issue of test size and power. We agree that a conservative test (one whose size can fall below alpha) may sacrifice power.
Our objective is distribution-free false-positive rate (FPR) control. That is, we wish to keep the FPR at or below alpha for every distribution of X and Y, because in our regime (nonstationary time series with few independent replicates) the scientist often cannot verify distributional assumptions. Inspired by the reviewer’s comment, we now show (new Proposition 14) that the perfect match probability can be made arbitrarily close to 1/n<sup>!</sup>. As a consequence, any reported perfect match p-value below 1/n<sup>!</sup> would break the distribution-free validity of the test.
A test that exploits distributional structure could likely access lower p-values; we have now explored how the empirical FPR of the permute-match test varies with the data-generating process (see our response to reviewer 2's recommendation 1 below).
(2) The computational time can be an issue for a moderately large sample size when calculating the X / Y-perfect match. It will be beneficial to include discussions on the implementations of the test.
We agree this is an important consideration. We have added the following text to the Discussion:
“The test appears computationally tractable for relevant sample sizes: Our implementation of the permute match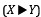 procedure completed a single test of dependence in the setting of Fig 2 with an average runtime of 3 seconds when n = 10 on a 2023 14-inch MacBook Pro with an M2 Pro processor and 16 GB RAM (see Source data 1). For n > 10, a standard permutation test already can report a p-value below 3 × 10<sup>−8</sup> so the perfect match test is likely unnecessary for typical applications.”
procedure completed a single test of dependence in the setting of Fig 2 with an average runtime of 3 seconds when n = 10 on a 2023 14-inch MacBook Pro with an M2 Pro processor and 16 GB RAM (see Source data 1). For n > 10, a standard permutation test already can report a p-value below 3 × 10<sup>−8</sup> so the perfect match test is likely unnecessary for typical applications.”
Recommendations for the authors:
Reviewer #1 (Recommendations for the authors):
A few more minor notes/comments:
As a personal preference, I like it when figures printed in grayscale retain their meaning (when possible). Just FYI, Figure 3D in grayscale is uninterpretable. Not saying a change is needed, just pointing it out.
We changed Fig 3D to address a comment above, and think the new version better distinguishes between the permute-match and permutation test results in greyscale.
On line 304, is that a lower bound or an upper bound? Maybe the issue is the probability mentioned on line 305 is not clear.
As this point is not the main focus of the investigation, we have rephrased it to make it less technical and eliminate the issue of which bound is in question.
“It seems likely that the tests could be further modified to report an even lower p-value when an X- and Y -perfect match occur simultaneously, as in Fig 3B. However, we have not investigated further and this problem is left for future efforts.”
I am not sure if this is a weakness, but the p-value changing depending on the choice of whether to apply the X-perfect match or Y-perfect match test first is fascinating. The authors did discuss this very issue at several points in the manuscript. It is slightly unsettling to me that there isn't an exact p-value for a given set of data. This one point gives me a new perspective on statistics.
In full transparency, I don't believe I have to background to thoroughly review the appendix. I did read through it and did not notice any errors, but I couldn't confidently say there are not any small mathematical errors or any logical flaws in the proofs. Some sections were not easy to follow (my own shortcomings, the writing appeared sufficient for more of an expert to understand).
We greatly appreciate the reviewer’s time and effort tackling an appendix outside their comfort zone.
Reviewer #2 (Recommendations for the authors):
(1) In the numerical experiment session, the authors should include the null situation, i.e., the performance of the test when X and Y are independent. This helps assess the size of the test.
We have added a section on size to our results section, copied below:
“The permute-match test’s false positive rate depends on the process tested. The permute-match test is conservative – meaning that its false positive rate can fall below the significance level – because both the permutation test and perfect match test are conservative. As discussed elsewhere [29], the permutation test is conservative when α is not one of its possible p-values and when ties may occur between the original correlation and shuffled correlations. Checking for a perfect match is similarly conservative. The actual probability of a false-alarm perfect match event can vary depending on the process being tested. To see this consider the permute-match test in the setting where n = 3, where α = 0.05, and where r<sub>X,Y</sub>= 0 (independent X and Y). Note that in this case, obtaining a Y -perfect match (and thus p = 1/n<sup>n</sup>) is necessary and sufficient to detect dependence since α is too low for detection by either the permutation test or the p = 2/n<sup>n</sup> leg of the permute-match test. In the linear system of Fig 2, we observed among 5000 simulations a detection rate of 0.0148, significantly below the upper bound of 1/3<sup>3</sup> (Figure 2 - Source data 1; one-tailed exact binomial test, p < 10<sup>−20</sup>). Conversely, in the nonlinear system of Fig S2, this same event (Y -perfect match under n = 3 and r<sub>X,Y</sub> = 0) occurs with a detection rate of 0.0328, not significantly 9 below the upper bound of 1/3<sup>3</sup> (Figure S2 - Source data 1; one-tailed exact binomial test, p = 0.059). Thus, depending on the underlying process studied, the actual chance of a perfect match happening under independence may be near or significantly below the theoretical upper bound.”
(2) Some insights regarding the choice of rho should be provided. Especially, are there any examples that the classical test, such as the Pearson correlation or Granger causality test does not work?
We have redone the example of Appendix 4 with Pearson correlation, showing that Pearson correlation has substantially lower power than cross-map skill in this case (compare figures S2 and S3).
(3) Line 34 - 35, page 2: Correlations and causality should be separately considered. This sentence talks more about causality rather than correlation.
We appreciate the reviewer’s perspective and agree that correlation and causality are distinct.
We feel that pointing out the issue of spurious correlations is helpful to orient our readers, especially those from a broad scientific audience. In the text, we define “correlation” as a descriptive statistic (rather than normalized covariance), and later distinguish it from “dependence”, which has causal implications due to Reichenbach’s common cause principle. We believe this distinction provides a useful backdrop for practitioners who use statistical methods but are perhaps new to thinking deeply about statistical dependence.
(4) Please add some discussions on the situation that X_i depends on Y_{i - j} for some j > 0, which is associated with the setting of Granger causality test.
We have added the following to the discussion:
“No distributional assumptions are required, and the correlation function ρ can be completely arbitrary. For instance, ρ could include a lag to detect delayed dependence, or even evaluate the correlation strength at several lags and report the strongest among them [39].”
le doux commerce
“Le doux commerce” 可翻译为中文:
“温和的商业”、“温柔的贸易”,或更符合思想史语境的译法:“商业的柔化作用”、“温和商业论”。
其含义是:
“温和商业”指启蒙时代的一种思想观点:贸易和经济交换能够使社会变得更加和平、文明与合作。因为人们在商业往来中形成相互依赖的关系,因此更有动力避免冲突,通过谈判和交换来解决问题。
这一概念通常与法国思想家 孟德斯鸠(Montesquieu)联系在一起。他在《论法的精神》(1748)中提出,商业能够“软化风俗”,促进不同民族之间的理解与交往。
后来,阿尔贝·赫希曼(Albert Hirschman)对这一观点进行了分析和批判,指出商业确实可能带来合作、理性和文明化,但也可能产生新的不平等和道德问题。
因此,“le doux commerce”通常包含以下几个层面的意思:
贸易代替冲突:经济利益促使人们通过协商而非暴力解决分歧;
利益促进合作:个人追求自身利益的过程,也可能创造相互依存;
交往培养文明:频繁的商业接触让陌生人之间形成信任和礼仪。
不过,它并不是说商业必然带来和平,而是一种启蒙时期关于市场、文明与社会进步关系的理想化观点。现代研究也强调,商业既可能促进和平与开放,也可能加剧不平等、剥削或竞争。
简而言之:
“Le doux commerce” = “温和商业”这一启蒙思想,即认为商业交换能够驯化人性、改善社会关系,并使人类从冲突走向合作。
programming-in-language
oh i remember james talking to me about this huh.
i wonder if it is just abstractions all the way up forever
Notable studentsYang Zhilin
oh hes had a lot more i think
Founders / Founding Team Members
Devendra Chaplot @dchaplot PhD, Founding Member Thinking Machines / Mistral Zhilin Yang PhD, Founder & CEO, Moonshot AI Jimmy Ba @jimmybajimmyba MSc/PhD, Co-founder xAI Hubert Tsai PhD, Co-founder Spuree, Apple Nitish Srivastava @nitishsr PhD, Co-founder Perceptual Machines; Co-founder Vayu Robotics Charlie Tang PhD, Co-founder Perceptual Machines, DE Shaw.
Professors Paul Liang @pliang279 PhD, MIT Ben Eysenbach @ben_eysenbach PhD, Princeton University Ruosong Wang @RuosongW PhD, Peking University Bhuwan Dhingra @bhuwandhingra PhD, Duke University Roger Grosse @RogerGrosse Postdoc, University of Toronto Alexander Schwing Postdoc, UIUC
Research Scientists Shuyan Zhou @shuyanzh36 , Postdoc, Meta Superintelligence Lab Tiffany Min @SoYeonTiffMin PhD, Microsoft AI Murtaza Dalal @mihdalal PhD, Tesla AI Minji Yoon @MinjiYoon90 , PhD, Microsoft AI Shrimai Prabhumoye PhD, NVIDIA AI, Mistral Haitian Sun @sun_haitian PhD, Google DeepMind Emilio Parisotto PhD, Google DeepMind Lisa Lee PhD @rl_agent , Google DeepMind Manzil Zaheer @ManzilZaheer PhD, Google DeepMind Jamie Kiros PhD, Google Brain, OpenAI Yuri Burda PhD, OpenAI, Anthropic Cody Severinski PhD, Amazon
In March 2023, Yang co-founded Moonshot AI with Zhou Xinyu and Wu Yuxin, his Splay band-mates and classmates at Tsinghua. The company name comes from the album The Dark Side of the Moon and the company was launched on its 50th anniversary.[1][2][3]
this is cool. very interesting naming method.
i might do that.
Bestuurders die in een jaar tijd te veel strafpunten verzamelen vanwege overtredingen of misdrijven worden extra in de gaten gehouden.
En verdere consequenties?
appen achter het stuur
met handen toch?
My parents crossed fingers so he’d never come back, lit novena candles so he would.
This might also mean they are possessed by a ghost, as one could not have two contridicting opinion
eLife Assessment
This important study by Otgonbaatar and colleagues employs advanced live microscopy, optogenetics, and an endogenous fluorescent timer system to investigate short- and long-term stabilization dynamics of β-catenin/Armadillo (Arm) during Drosophila development. The authors identify an unexpected and functionally relevant enrichment of stabilized junctional Arm in leading-edge cells during dorsal closure, providing evidence for a stabilization mechanism that appears independent of canonical Wingless signaling. These findings are significant because they expand current understanding of β-catenin/Arm beyond its canonical signaling functions and suggest a role in tissue mechanics and force transmission during dorsal closure. The proposed model represents a key advance in the field, but the strength of evidence is currently incomplete: the main conclusions regarding Wingless independence, JNK-mediated regulation, and the mechanical role of stabilized Arm are only partially supported by the available data and would benefit from further experimental testing and corroboration.
Reviewer #1 (Public review):
In this study, Otgonbaatar and colleagues investigate the stability of Armadillo (Arm) during Drosophila development using a creative tandem fluorescent protein timer approach via endogenous tagging of Arm. The tagging strategy allows for newly synthesised and longer-term stabilised Arm pools to be distinguished from one another. Specifically, the authors address the functional relevance of and mechanism behind the stabilisation of junctional Arm during dorsal closure.
The authors show that Arm is stabilised at the leading edge during dorsal closure. Using a sophisticated optogenetics approach, which allows for acute perturbations, they show that stabilised Arm is functionally required for dorsal closure. Increasing Wg (by overexpression) did not affect dorsal closure or Arm stability, in contrast to Axin overexpression, which reduces Wg/Arm signalling. In line with canonical signalling control of Arm levels being critical, stabilisation of Arm by N-terminal mutations disrupted dorsal closure. However, the same deletion is also expected to affect interaction with alpha-catenin. Co-localisation with E-cadherin and actin suggests a junctional role of leading-edge localised Arm. Optogenetic targeting of alpha-catenin points towards a key role of adherence junctions in dorsal closure. Allele replacement with mutant variants of Arm to affect adherence junction complex assembly further indicates an important contribution of coupling between Arm and alpha-catenin. Using overexpression approaches, the authors suggest that Dsh and Jnk contribute to dorsal closure.
This microscopy- and optogenetics-based study is generally well-conducted and provides strong evidence for stabilised Arm during dorsal closure, as well as its functional importance. This is an important discovery relevant to morphogenesis and potentially mechanotransduction. From a technical perspective, the validated beta-catenin timer provides a valuable tool for the field. The timer has revealed that Arm stabilisation does not coincide with Wg stripes, suggesting a Wg-independent stabilisation mechanism that may instead depend on adherence junction assembly, especially the interaction of Arm with alpha-catenin. However, as N-terminal deletion within Arm and Axin overexpression also disrupted dorsal closure, substantial ambiguity remains. Can suppression of the beta-catenin degradation machinery be ruled out as a regulatory mechanism? An expansion of ArmTimer mutant variants could contribute to testing the authors' conclusion further. Structural insights into junctional interactions involving Arm (e.g., 10.1074/jbc.M114.554709) could, for example, be used for further functional exploration by mutagenesis. The direct mechanistic impact of JNK and its potential link to Dsh in dorsal closure remains less compelling.
In summary, this is a highly relevant and important study, potentially pointing to a novel stabilisation mechanism of beta-catenin in development. Further corroboration of the mechanism, to test whether it is indeed distinct from canonical signalling, would be needed to support the conclusions.
Reviewer #2 (Public review):
Summary:
Otgonbaatar et al. sought to investigate β-catenin/Arm protein lifetime and stabilization dynamics in vivo during embryonic development. To address this question, the authors developed an endogenous tandem fluorescent protein timer (tFP) system that enables the visualization of newly synthesized versus long-lived Arm protein in vivo. Using this approach, the authors sought to determine where stabilized Arm accumulates during development and how it contributes to dorsal closure.
Strengths:
A major strength of the study is the development and application of the endogenous Arm timer system, which provides a powerful approach for monitoring protein stabilization dynamics in living tissues. Using this system, the authors unexpectedly found that the strongest Arm stabilization occurs not in Wnt signaling regions, but at the leading edge cells during dorsal closure. The study combines quantitative live imaging, optogenetic perturbation, genetic analysis, and structure-function approaches to demonstrate that stabilized junctional Arm interacts with α-catenin and contributes to tissue mechanics required at the leading edge for dorsal closure. Particularly compelling is the combination of multiple perturbations, including optogenetic disruption of Arm or α-catenin, Axin overexpression, and Arm mutants, which produce consistent dorsal closure defects.
Some conclusions are generally supported by the presented data. The work provides strong evidence that Arm plays an important role in dorsal closure. The identification of a requirement for the Dishevelled DEP domain and JNK signaling supports a non-canonical regulatory mechanism controlling dorsal closure.
Weaknesses:
(1) Conclusions are made regarding force transmission;(however, no experimental evidence is provided to support these conclusions.
(2) The conclusion was made that Wingless does not affect dorsal closure. However, this was based solely on Wingless overexpression in the amnioserosa, and the level of Wingless expression was not quantified. One possibility is that this level was not sufficient to see an effect. Alternatively, Wingless may have a role in migrating epithelium rather than the amnioserosa. Indeed, it is known that wingless mutants display a defect in dorsal closure.
(3) The effect of JNK knockdown on Arm localization maybe is indirect, and due to a secondary consequence on disruption of epithelial morphology rather than a direct effect of JNK on Arm.
(4) Some conclusions rely on overexpression-based perturbations (e.g., Axin or Arm mutants), which may not fully recapitulate endogenous physiological regulation.
(5) The Arm timer was not able to detect Wingless-dependent Arm stabilization in stripes. This finding demonstrates that the timer is not sensitive enough to thoroughly analyze Arm dynamics.
Overall, this work provides important conceptual advances in understanding junctional β-catenin/Arm function during dorsal closure. The endogenous fluorescent timer approach will likely be broadly useful to the community for studying protein stability dynamics in vivo, and the findings expand current views of β-catenin by highlighting its mechanical and junctional functions during tissue morphogenesis.
Author response:
We are glad the reviewers found the tandem fluorescent timer approach valuable and the leading-edge Arm stabilization finding significant.
We agree with the Assessment that our evidence for three specific claims Wingless-independence, JNK-mediated regulation of Arm stability, and a direct mechanical/force-transmission role for stabilized Arm is currently incomplete, and we will revise the text throughout to reflect this more precisely rather than overstating the current data. In addition, we commit to two new experiments, both using existing reagents and fly stocks, that speak directly to the two most experimentally tractable points raised by the reviewers:
(1) Re-staining our existing JNK-RNAi and JNK-overexpression embryos for E-cadherin (reagent already validated in Figure 4), to test whether JNK acts directly on junctional architecture or only indirectly, via broader epithelial disruption.
(2) Imaging ArmTimer in a wingless loss-of-function background, to directly test Wingless-dependence of leading-edge Arm stabilization as the reciprocal of our existing overexpression data.
We address each public review point below and outline the accompanying text revisions.
On Wingless independence (Reviewer #1; Reviewer #2, Weaknesses #2 and #5; Recommendation #1):
We agree that our current evidence unquantified Wg overexpression restricted to the amnioserosa (C381-Gal4) and uniform overexpression, alongside the absence of detectable Wg-stripe-associated Arm-Timer signal supports a more limited conclusion than "Wingless-independent" as currently stated. We will revise our language throughout the Abstract, Results, and Discussion to state that canonical Wg overexpression does not detectably enhance leading-edge Arm stabilization or perturb dorsal closure under our conditions, rather than asserting pathway independence. As noted above, we commit to imaging ArmTimer in a wg mutant background to test this directly, complementing our overexpression data with the reciprocal loss-of-function manipulation.
On the related point that the Timer's failure to detect a Wg-stripe-associated stabilization signal could reflect a sensitivity limitation rather than a true absence of stabilization (Reviewer #2, Weakness #5): we agree and will state this explicitly rather than treating absence of signal as evidence of absence. This does not undermine the positive leading-edge finding, which is not defined relative to the stripe comparison: all embryos, channels, and time points were imaged and rendered using identical laser power and brightness/sensitivity settings, and the leading-edge RFP signal clearly exceeds background under those same acquisition conditions. We also note that detection limits of this kind are a recognized challenge for endogenously tagged reporters of canonical Wnt/β-catenin signaling generally, including in mammalian systems, and cite two studies already in our bibliography that report the same class of limitation: de Man et al. (2021, eLife 10:e66440) and Ambrosi et al. (2022, eLife 11:e64498). We have added this clarification, with these citations, to the Results (paragraph describing Figure 3).
On the mechanistic link between Arm stability and destruction-complex activity (Reviewer #1):
We agree that because both ΔArm and Axin overexpression converge on the destruction complex, our data cannot yet fully separate "escape from degradation" from "impaired α-catenin/junctional coupling" as the operative mechanism. We will revise the Discussion to state this ambiguity explicitly and will treat the ArmTimer-AA result (partial α-catenin-binding disruption via phosphosite mutation, independent of destruction-complex regulation) as the strongest current evidence isolating the junctional-coupling mechanism. We also thank Reviewer 1 for pointing us to Pokutta, Choi, Ahlsen, Hansen & Weis (2014, J Biol Chem 289:13589-13601), which structurally and thermodynamically characterized the mammalian cadherin·β-catenin·α-catenin complex and showed that α-catenin binding to β-catenin is a distinct, allosterically regulated interface cadherin binding increases β-catenin's affinity for α-catenin roughly 10-fold, and α-catenin homodimerization independently competes with β-catenin binding. We have added this citation to the Discussion as structural support for treating cadherin engagement, α-catenin coupling, and destruction-complex regulation as mechanistically separable interfaces, and note that the crystallized β-catenin·α-catenin interface provides a structural template for future experiments for example, structure-guided point mutations at the homologous interface residues in Arm, or in vitro binding assays comparing wild-type and threonine-mutant (T111A/T121A) Arm affinity for α-catenin.
We do not, however, believe a destruction-complex-independent stabilizing allele of Arm is a tractable experiment to close this gap directly: any allele that stabilizes Arm without engaging the destruction complex is, by definition, a Wnt pathway gain-of-function allele, since destruction-complex-mediated degradation is the very regulatory step that canonical Wnt signaling controls. Nor would restricting the allele to a transcriptionally inactive form of Arm cleanly resolve the confound: Wnt/TCF target loci include dedicated repressive TCF-binding sites (Blauwkamp, Chang & Cadigan, 2008, EMBO J 27:1436-1446), so a transcriptionally "dead" stabilized Arm could still alter transcription by disrupting TCF-mediated repression. We therefore treat this as a genuine, currently unresolvable confound of the overexpression approach, and rely instead on the CRY2 optogenetic and ArmTimer-AA results as the strongest available evidence isolating a junctional-coupling contribution. We have added this reasoning, with both citations, to the Discussion.
On JNK acting on Arm directly vs. indirectly (Reviewer #1; Reviewer #2, Weakness #3 and Recommendation #2):
This is the most actionable point raised by both reviewers. As noted above, we commit to re-imaging and re-staining our existing JNK-RNAi and JNK-overexpression embryos for E-cadherin to determine whether junctional/polarity architecture is broadly disrupted under these conditions (indirect mechanism) or whether E-cadherin localization is comparatively preserved while Arm stabilization is specifically altered (direct mechanism). In the meantime, we note that a direct mechanism is biochemically plausible: in mammalian cells, JNK phosphorylates β-catenin directly and regulates adherens junction integrity, and JNK activity separately controls the binding of α-catenin to the junctional complex (Lee, Koria, Qu & Andreadis, 2009, FASEB J 23:3874-3883; Lee, Padmashali, Koria & Andreadis, 2011, FASEB J 25:613-623). We cite these as precedent that a direct route from JNK to junctional β-catenin/α-catenin regulation exists in another system, while being explicit that this does not establish the same mechanism in Drosophila dorsal closure that will be tested directly by the E-cadherin re-staining experiment. We have added these citations and this caveat to the Discussion.
On the Dsh-DEP-to-JNK mechanistic link (Reviewer #1, Public Review #2 and Recommendation #5):
We agree that our data show the Dsh-DEP requirement and the JNK requirement for dorsal closure as parallel, independent findings rather than a demonstrated linear pathway in our system. To provide context for why we consider a DEP-to-JNK connection a reasonable working hypothesis, we searched the literature in both Drosophila and vertebrates and will cite six additional studies establishing this link: Axelrod et al. (1998) and Axelrod (2001), establishing that DEP-dependent membrane recruitment and unipolar localization of Dishevelled are specifically required for planar polarity signaling, distinct from Wingless signaling; Paricio et al. (1999) and Fanto et al. (2000), showing Dishevelled acts through Misshapen and Rac1/RhoA to the same JNK module used in dorsal closure; and Moriguchi et al. (1999) and Yamanaka et al. (2002), showing biochemically in vertebrates that the DEP domain of Dvl-1 selectively activates JNK independent of β-catenin/TCF-LEF activity, and that this JNK requirement is conserved in Xenopus convergent extension, the vertebrate process most functionally analogous to dorsal closure. We will state explicitly that this precedent, while now cross-species, comes from planar-cell-polarity and convergent-extension assays rather than dorsal closure itself, so it supports the plausibility of a Dsh/Dvl-DEP-to-JNK connection without establishing that the identical pathway operates in our system.
On the Dsh DIX/DEP domain-separability argument (Reviewer #1, Recommendation #4):
We thank the reviewer for pointing us to Gammons, Renko, Johnson, Rutherford & Bienz (2016, Mol Cell 64:92-104), which showed that the Wnt signalosome itself is assembled by head-to-tail DEP domain swapping between Dishevelled molecules, and that this DEP-dependent oligomerization is directly required for canonical Wnt pathway activity not restricted to the non-canonical/planar-polarity branch as we had implied. We agree this evidence undercuts our previous interpretation of the DshΔDEP dorsal closure phenotype as evidence for a strong non-canonical/polarity-specific role for the DEP-dependent branch of Dsh. We have revised the Discussion accordingly: we now state that the DEP domain is required for the morphogenetic program culminating in dorsal closure, cite Gammons et al. directly, and note that this requirement does not by itself establish a non-canonical/polarity-specific role, since we cannot rule out a contribution from DEP-dependent canonical Wnt signalosome assembly.
On force transmission (Reviewer #2, Weakness #1):
We agree that we have not directly measured force or tension at the leading edge, and that our current data (colocalization with actin/E-cadherin, and functional requirement shown via CRY2 optogenetics and mutant analysis) are consistent with, but do not directly demonstrate, a role in force transmission. We do not have the in-house expertise to perform direct force/tension measurements (e.g., laser ablation, junctional tension assays), so we will not be adding such an experiment in this revision. Instead, we have revised the language throughout the manuscript including two Discussion section headings that previously stated a mechanical role for stabilized Arm as established fact to consistently present the mechanical/force-transmission role as a hypothesis raised by our data, not a demonstrated conclusion, and we retain a clear statement that direct force measurement (ideally in collaboration with groups with the relevant biophysical expertise) is future work rather than a claim we are making in this manuscript.
On the phosphomimetic threonine mutant (Reviewer #1, Recommendation #2):
ArmTimer-AA (T111A, T121A) was generated with the expectation that the tyrosine phosphosite mutants (ArmTimer-EE, ArmTimer-FF) would be the primary drivers of any dorsal closure phenotype, given their proposed role in E-cadherin binding; the pronounced zippering defect we observed in ArmTimer-AA was therefore an unanticipated finding rather than a predicted result. We have not generated the reciprocal phosphomimetic ArmTimer-EE(Thr) (T111E, T121E) allele. Generating and characterizing this allele is a substantial undertaking we estimate over a year including allele generation, validation, and phenotypic characterization and we will state this explicitly in the Discussion as planned future work rather than part of the current revision.
On confirmation of myristoylated-Dsh membrane targeting (Reviewer #1, Recommendation #3):
We cannot confirm that myristoylation localizes all Dsh protein to the membrane. However, this strategy has extensive prior genetic validation using the identical Src-derived myristoylation sequence: it was originally used to tether Armadillo and shown sufficient for constitutive Wnt pathway activation (Zecca, Basler & Struhl, 1996; Tolwinski & Wieschaus, 2001, 2004), and the same approach was subsequently applied to GSK3 and Dishevelled, in each case producing the expected pathway-activation phenotypes (Mannava & Tolwinski, 2015; Kaur et al., 2017). We have added these citations to the Results where the Myr-Dsh constructs are introduced.
On overexpression-based perturbations versus endogenous regulation (Reviewer #2, Weakness #4):
We would like to clarify that most of the Arm alleles used in this study including all of the point-mutant Timer alleles (ArmF1a, ArmTimer-FF, ArmTimer-EE, ArmTimer-AA) central to our mechanistic conclusions were generated as knock-ins at the endogenous ‘arm’ locus via MiMIC/RMCE, not overexpressed. The two exceptions are ΔArm and ArmS56A, expressed from UAS constructs because both are gain-of-function alleles anticipated to be lethal if expressed from the endogenous locus, based on prior experience with similarly stabilizing mutations. Axin overexpression was used because no Axin mutant or knock-in allele was generated for this study; we agree an endogenous Axin allele would be the ideal complement and will state this explicitly as a limitation, while noting that our CRY2 optogenetic perturbations of Arm and α-catenin which act acutely on the endogenous proteins provide an orthogonal line of evidence supporting the same conclusions. We have added this clarification to the Discussion.
not being forced to think anymore
AI depriving people the capacity to think deeply
eLife Assessment
This study presents a valuable RNA velocity solution which integrates cell differentiation and gene regulation, with a balance between neuralODE and raw gene space. The evidence supporting the claims of the authors is solid, although inclusion of discussion on the challenges in capturing cell cycle transitions would have strengthened the study. The work will be of interest to scientists working in the field of computational biology and gene regulation.
Reviewer #1 (Public review):
Summary:
In the paper, the authors propose a new RNA velocity method, TSvelo, which predicts the transcription rate linearly based on the expression of RNA levels of transcription factors. This framework is an extension of its recent work TFvelo by including unspliced reads and designing a coherent neuralODE framework. Improved performance was demonstrated in six diverse datasets.
Strengths:
Overall, this method introduces innovative solutions to link cell differentiation and gene regulation, with a balance between model complexity (neuralODE) and interpretability (raw gene space).
Comments on revised version:
I thank the authors for further revision, and I do not have any other concerns. I believe it is an important contribution to this field of trajectory inference and gene regulation.
Reviewer #3 (Public review):
Despite the abundance of RNA velocity tools, there are still major limitations, and there is strong skepticism about the results these methods lead to. In this paper, the authors try to address some limitations of current RNA velocity approaches by proposing a unified framework to jointly infer transcriptional and splicing dynamics. The method is then benchmarked on 6 real datasets against the most popular RNA velocity tools.
Comments on revised version:
The Authors addressed my 2 follow-up comments suitably.
Thanks for the time you took addressing them. I have no further comments.
Author response:
The following is the authors’ response to the previous reviews
Public Reviews:
Reviewer #1 (Public review):
Summary:
In the paper, the authors propose a new RNA velocity method, TSvelo, which predicts the transcription rate linearly based on the expression of RNA levels of transcription factors. This framework is an extension of its recent work TFvelo by including unspliced reads and designing a coherent neuralODE framework. Improved performance was demonstrated in six diverse datasets.
Strengths:
Overall, this method introduces innovative solutions to link cell differentiation and gene regulation, with a balance between model complexity (neuralODE) and interpretability (raw gene space).
Comments on revised version:
The authors have added comprehensive analyses in this revision, and all of my concerns have been very well addressed. Here, I just want to re-emphasize the original points 1 and 3.
(1) The analysis and clarification are very helpful - thanks! I found that Fig. R1 and R2 are very insightful, as DoRothEA-only returns much worse performance. Please consider adding these two figures to the supp figure and possibly highlighting your setting for edge pruning (down-weights); therefore, the model is more likely to be affected by false negatives than false positives in the TF-target prior.
We thank the reviewer for the positive feedback and for recognizing the value of the additional analyses. We have added the previous Fig. R1 and Fig. R2 to the Supplementary Information as Fig. S13 and Fig. S14, respectively, and have referred to them in the revised manuscript.
We have also expanded the description of the TF–target prior used in TSvelo in the “Acquiring Prior Knowledge of Gene Regulatory Relations” subsection of the Methods. As noted by the reviewer, TSvelo is expected to be less sensitive to false-positive TF–target interactions because unsupported edges can be down-weighted during training. In contrast, missing true regulatory interactions are not represented in the prior network and therefore cannot contribute to the learned regulatory dynamics, making the model potentially more sensitive to false negatives.
(3) Please consider adding some discussion on the challenges in capturing cell cycle transitions.
We thank the reviewer for this suggestion. We have added a brief discussion in the Discussion section on the challenges of modeling cell-cycle transitions. In particular, cell-cycle progression is often characterized by cyclic dynamics and overlapping transcriptional programs, which can complicate the inference of directional state transitions and regulatory relationships.
Reviewer #3 (Public review):
Despite the abundance of RNA velocity tools, there are still major limitations, and there is strong skepticism about the results these methods lead to. In this paper, the authors try to address some limitations of current RNA velocity approaches by proposing a unified framework to jointly infer transcriptional and splicing dynamics. The method is then benchmarked on 6 real datasets against the most popular RNA velocity tools.
Comments on revised version.
The Authors addressed all my comments suitably. I'd like to thank them for the time they spent addressing them: the revised paper is much more convincing.
I have 2 very minor follow-up concerns:
(1) I appreciated the simulation study, however, no null simulation is present.
We know RNA velocity tools are inclined to provide false positives: trajectories even when the data doesn't have any.
I'd be helpful to add null simulations where the data has no trajectories and see if methods erroneously identify any.
We thank the reviewer for this helpful suggestion. We have added null simulations to evaluate TSvelo and baseline approaches on data without underlying dynamic structure. Specifically, we generated a null dataset including 200 genes and 600 cells by independently sampling spliced (S) and unspliced (U) counts, thereby removing any coherent transcriptional relationship between them.
When applying scVelo and UniTVelo to this data, no genes passed the velocity gene selection step under the default likelihood-based filtering, and no velocity field could be obtained. We further tested TSvelo, Dynamo, and cellDancer on the same null data and observed that all three methods still produce trajectory-like patterns despite the absence of true dynamics (See Supplementary Information as Fig. S17).
Including TSvelo, many RNA velocity and trajectory inference approaches assume that they are applied to datasets reflecting underlying dynamic biological processes. We agree that incorporating additional checks during preprocessing could help prevent applying velocity analysis to non-dynamic datasets. We have added this discussion to the revised manuscript.
(2) Several of the novel analyses are only reported in the Supplementary material and only references in the main text (e.g., "A validation of TSvelo on simulated data is provided in Fig. S1 and Fig. S2 in the Supplementary Information."). This is pity!
If allowed, I'd add some comments about the new analyses (simulations, computational benchmarks, etc...) also in the main text.
We thank the reviewer for this suggestion. We agree that several analyses presented in the Supplementary Information provide important support for our conclusions. To improve their visibility, we have expanded the corresponding descriptions in the main text and briefly summarized the key findings of the relevant Supplementary Figures instead of only citing them. These revisions have been made for Fig. S1, Fig. S2, Fig. S10, Fig. S12, Fig. S13, Fig. S14 and Fig. S17. In particular, we have incorporated a summary of the simulation results at the end of the subsection “Estimate RNA Velocity with TSvelo” in the Results section, and added a discussion of the computational benchmarking analyses in the Discussion section. We hope these changes improve the accessibility of these results while maintaining a concise presentation of the main findings.
Recommendations for the authors:
Reviewer #3 (Recommendations for the authors):
I suggest the paper to undergo (very) minor revisions as detailed in the Public Review.
Simone Tiberi, The University of Bologna
We sincerely thank all reviewers for their thoughtful suggestions, which have helped improve the clarity and overall presentation of the manuscript.
RRID: AB_2941781
DOI: 10.3389/fendo.2026.1879250
Resource: (Siemens Cat# 10995541, RRID:AB_2941781)
Curator: @evieth
SciCrunch record: RRID:AB_2941781
RRID: AB_2909501
DOI: 10.3389/fendo.2026.1879250
Resource: (Siemens Cat# 03649928, RRID:AB_2909501)
Curator: @evieth
SciCrunch record: RRID:AB_2909501
RRID:AB_2909499
DOI: 10.3389/fendo.2026.1879250
Resource: (Siemens Cat# 02230141, RRID:AB_2909499)
Curator: @scibot
SciCrunch record: RRID:AB_2909499
RRID:AB_2941780
DOI: 10.3389/fendo.2026.1879250
Resource: (Siemens Cat# 10995628, RRID:AB_2941780)
Curator: @scibot
SciCrunch record: RRID:AB_2941780
BL3642
DOI: 10.1371/journal.pgen.1011475
Resource: RRID:BDSC_3642
Curator: @bdscstockkeepers
SciCrunch record: RRID:BDSC_3642
BL7013
DOI: 10.1371/journal.pgen.1011475
Resource: RRID:BDSC_7013
Curator: @bdscstockkeepers
SciCrunch record: RRID:BDSC_7013
BL52676
DOI: 10.1371/journal.pgen.1011475
Resource: RRID:BDSC_52676
Curator: @bdscstockkeepers
SciCrunch record: RRID:BDSC_52676
BL80428
DOI: 10.1371/journal.pgen.1011475
Resource: RRID:BDSC_80428
Curator: @bdscstockkeepers
SciCrunch record: RRID:BDSC_80428
BL34685
DOI: 10.1371/journal.pgen.1011475
Resource: RRID:BDSC_34685
Curator: @bdscstockkeepers
SciCrunch record: RRID:BDSC_34685
BL34617
DOI: 10.1371/journal.pgen.1011475
Resource: RRID:BDSC_34617
Curator: @scibot
SciCrunch record: RRID:BDSC_34617
BL6357
DOI: 10.1371/journal.pgen.1011475
Resource: RRID:BDSC_6357
Curator: @scibot
SciCrunch record: RRID:BDSC_6357
BL35741
DOI: 10.1371/journal.pgen.1011475
Resource: RRID:BDSC_35741
Curator: @scibot
SciCrunch record: RRID:BDSC_35741
BL5905
DOI: 10.1371/journal.pgen.1011475
Resource: RRID:BDSC_5905
Curator: @scibot
SciCrunch record: RRID:BDSC_5905
BL9146
DOI: 10.1371/journal.pgen.1011475
Resource: RRID:BDSC_9146
Curator: @scibot
SciCrunch record: RRID:BDSC_9146
BL30557
DOI: 10.1371/journal.pgen.1011475
Resource: RRID:BDSC_30557
Curator: @scibot
SciCrunch record: RRID:BDSC_30557
BL8530
DOI: 10.1371/journal.pgen.1011475
Resource: RRID:BDSC_8530
Curator: @scibot
SciCrunch record: RRID:BDSC_8530
BL458
DOI: 10.1371/journal.pgen.1011475
Resource: RRID:BDSC_458
Curator: @scibot
SciCrunch record: RRID:BDSC_458
BL14492
DOI: 10.1371/journal.pgen.1011475
Resource: RRID:BDSC_14492
Curator: @scibot
SciCrunch record: RRID:BDSC_14492
AB_2273020
DOI: 10.1186/s12951-026-04390-6
Resource: (Proteintech Cat# 10366-1-AP, RRID:AB_2273020)
Curator: @evieth
SciCrunch record: RRID:AB_2273020
AB_2082037
DOI: 10.1186/s12951-026-04390-6
Resource: (Proteintech Cat# 14695-1-AP, RRID:AB_2082037)
Curator: @evieth
SciCrunch record: RRID:AB_2082037
AB_2263076
DOI: 10.1186/s12951-026-04390-6
Resource: (Proteintech Cat# 10494-1-AP, RRID:AB_2263076)
Curator: @evieth
SciCrunch record: RRID:AB_2263076
AB_2105691
DOI: 10.1186/s12951-026-04390-6
Resource: (Proteintech Cat# 15613-1-AP, RRID:AB_2105691)
Curator: @evieth
SciCrunch record: RRID:AB_2105691
CVCL_0145
DOI: 10.1186/s12951-026-04390-6
Resource: (ATCC Cat# CRL-2302, RRID:CVCL_0145)
Curator: @scibot
SciCrunch record: RRID:CVCL_0145
AB_10733242
DOI: 10.1186/s12951-026-04390-6
Resource: (Proteintech Cat# 21773-1-AP, RRID:AB_10733242)
Curator: @scibot
SciCrunch record: RRID:AB_10733242
RRID:SCR_015687
DOI: 10.1186/s12951-026-04390-6
Resource: DESeq2 (RRID:SCR_015687)
Curator: @scibot
SciCrunch record: RRID:SCR_015687
AB_10646467
DOI: 10.1186/s12951-026-04390-6
Resource: (Proteintech Cat# 19999-1-AP, RRID:AB_10646467)
Curator: @scibot
SciCrunch record: RRID:AB_10646467
CVCL_F0BB
DOI: 10.1186/s12951-026-04390-6
Resource: RRID:CVCL_F0BB
Curator: @scibot
SciCrunch record: RRID:CVCL_F0BB
SCR_016323
DOI: 10.1186/s12951-026-04390-6
Resource: StringTie (RRID:SCR_016323)
Curator: @scibot
SciCrunch record: RRID:SCR_016323
SCR_015530
DOI: 10.1186/s12951-026-04390-6
Resource: HISAT2 (RRID:SCR_015530)
Curator: @scibot
SciCrunch record: RRID:SCR_015530
SCR_011841
DOI: 10.1186/s12951-026-04390-6
Resource: cutadapt (RRID:SCR_011841)
Curator: @scibot
SciCrunch record: RRID:SCR_011841
SCR_013672
DOI: 10.1186/s12951-026-04390-6
Resource: ZEISS ZEN Microscopy Software (RRID:SCR_013672)
Curator: @scibot
SciCrunch record: RRID:SCR_013672
JAX:000664
DOI: 10.1186/s12951-026-04390-6
Resource: RRID:IMSR_JAX:000664
Curator: @scibot
SciCrunch record: RRID:IMSR_JAX:000664
SCR_002798
DOI: 10.1186/s12951-026-04390-6
Resource: GraphPad Prism (RRID:SCR_002798)
Curator: @scibot
SciCrunch record: RRID:SCR_002798
AB_2880408
DOI: 10.1186/s12951-026-04390-6
Resource: (Proteintech Cat# 26157-1-AP, RRID:AB_2880408)
Curator: @scibot
SciCrunch record: RRID:AB_2880408
SCR_002285
DOI: 10.1186/s12951-026-04390-6
Resource: Fiji (RRID:SCR_002285)
Curator: @scibot
SciCrunch record: RRID:SCR_002285
AB_3075517
DOI: 10.1186/s12951-026-04390-6
Resource: (ServiceBio Cat# GB11188, RRID:AB_3075517)
Curator: @scibot
SciCrunch record: RRID:AB_3075517
AB_2539916
DOI: 10.1186/s12951-026-04390-6
Resource: RRID:AB_2539916
Curator: @scibot
SciCrunch record: RRID:AB_2539916
AB_2181006
DOI: 10.1186/s12951-026-04390-6
Resource: (Abcam Cat# ab13826, RRID:AB_2181006)
Curator: @scibot
SciCrunch record: RRID:AB_2181006
AB_307284
DOI: 10.1186/s12951-026-04390-6
Resource: (Abcam Cat# ab9498, RRID:AB_307284)
Curator: @scibot
SciCrunch record: RRID:AB_307284
AB_2223009
DOI: 10.1186/s12951-026-04390-6
Resource: (Proteintech Cat# 14395-1-AP, RRID:AB_2223009)
Curator: @scibot
SciCrunch record: RRID:AB_2223009
AB_2082037
DOI: 10.1186/s12951-026-04390-6
Resource: (Proteintech Cat# 14695-1-AP, RRID:AB_2082037)
Curator: @scibot
SciCrunch record: RRID:AB_2082037
AB_1140040
DOI: 10.1186/s12951-026-04390-6
Resource: (Abcam Cat# ab6640, RRID:AB_1140040)
Curator: @scibot
SciCrunch record: RRID:AB_1140040
AB_10732601
DOI: 10.1186/s12951-026-04390-6
Resource: (Proteintech Cat# 20960-1-AP, RRID:AB_10732601)
Curator: @scibot
SciCrunch record: RRID:AB_10732601
AB_2139822
DOI: 10.1186/s12951-026-04390-6
Resource: (Proteintech Cat# 11257-1-AP, RRID:AB_2139822)
Curator: @scibot
SciCrunch record: RRID:AB_2139822
35249
DOI: 10.1126/sciadv.adn5417
Resource: RRID:BDSC_35249
Curator: @bdscstockkeepers
SciCrunch record: RRID:BDSC_35249
8605
DOI: 10.1126/sciadv.adn5417
Resource: RRID:BDSC_8605
Curator: @bdscstockkeepers
SciCrunch record: RRID:BDSC_8605
plasmid_17609
DOI: 10.1126/sciadv.adn5417
Resource: RRID:Addgene_17609
Curator: @scibot
SciCrunch record: RRID:Addgene_17609
RRID: IMSR_JAX:017535
DOI: 10.1038/s41592-026-03056-3
Resource: (IMSR Cat# JAX_017535,RRID:IMSR_JAX:017535)
Curator: @evieth
SciCrunch record: RRID:IMSR_JAX:017535
BDSC: 64349
DOI: 10.1016/j.xpro.2024.103456
Resource: RRID:BDSC_64349
Curator: @bdscstockkeepers
SciCrunch record: RRID:BDSC_64349
RRID:SCR_002798
DOI: 10.7554/eLife.108953
Resource: GraphPad Prism (RRID:SCR_002798)
Curator: @scibot
SciCrunch record: RRID:SCR_002798
RRID:AB_2576217
DOI: 10.7554/eLife.108953
Resource: (Thermo Fisher Scientific Cat# A-11034, RRID:AB_2576217)
Curator: @scibot
SciCrunch record: RRID:AB_2576217
RRID:Addgene_26924
DOI: 10.7554/eLife.108953
Resource: RRID:Addgene_26924
Curator: @scibot
SciCrunch record: RRID:Addgene_26924
RRID:AB_331623
DOI: 10.7554/eLife.108953
Resource: (Cell Signaling Technology Cat# 9242, RRID:AB_331623)
Curator: @scibot
SciCrunch record: RRID:AB_331623
RRID:AB_330288
DOI: 10.7554/eLife.108953
Resource: (Cell Signaling Technology Cat# 4967, RRID:AB_330288)
Curator: @scibot
SciCrunch record: RRID:AB_330288
RRID:AB_1904009
DOI: 10.7554/eLife.108953
Resource: RRID:AB_1904009
Curator: @scibot
SciCrunch record: RRID:AB_1904009
RRID:AB_303264
DOI: 10.7554/eLife.108953
Resource: (Abcam Cat# ab2739, RRID:AB_303264)
Curator: @scibot
SciCrunch record: RRID:AB_303264
RRID:AB_10706937
DOI: 10.7554/eLife.108953
Resource: (Cell Signaling Technology Cat# 5733, RRID:AB_10706937)
Curator: @scibot
SciCrunch record: RRID:AB_10706937
RRID:AB_443394
DOI: 10.7554/eLife.108953
Resource: (Abcam Cat# ab16502, RRID:AB_443394)
Curator: @scibot
SciCrunch record: RRID:AB_443394
RRID:AB_10859369
DOI: 10.7554/eLife.108953
Resource: (Cell Signaling Technology Cat# 8242, RRID:AB_10859369)
Curator: @scibot
SciCrunch record: RRID:AB_10859369
RRID:AB_675659
DOI: 10.7554/eLife.108953
Resource: (Santa Cruz Biotechnology Cat# sc-13119, RRID:AB_675659)
Curator: @scibot
SciCrunch record: RRID:AB_675659
RRID:AB_2533456
DOI: 10.7554/eLife.108953
Resource: (Thermo Fisher Scientific Cat# 40-2200, RRID:AB_2533456)
Curator: @scibot
SciCrunch record: RRID:AB_2533456
RRID:CVCL_0030
DOI: 10.7554/eLife.108953
Resource: (ICLC Cat# HTL95023, RRID:CVCL_0030)
Curator: @scibot
SciCrunch record: RRID:CVCL_0030
RRID:AB_306848
DOI: 10.5812/ijpr-170783
Resource: (Abcam Cat# ab8898, RRID:AB_306848)
Curator: @scibot
SciCrunch record: RRID:AB_306848
RRID:AB_2751009
DOI: 10.5812/ijpr-170783
Resource: (Abcam Cat# ab176880, RRID:AB_2751009)
Curator: @scibot
SciCrunch record: RRID:AB_2751009
RRID:AB_2295074
DOI: 10.5812/ijpr-170783
Resource: (Millipore Cat# 06-598, RRID:AB_2295074)
Curator: @scibot
SciCrunch record: RRID:AB_2295074
RRID:AB_2115283
DOI: 10.5812/ijpr-170783
Resource: (Millipore Cat# 06-599, RRID:AB_2115283)
Curator: @scibot
SciCrunch record: RRID:AB_2115283
RRID:CVCL_1G42
DOI: 10.1371/journal.pone.0348959
Resource: (RRID:CVCL_1G42)
Curator: @scibot
SciCrunch record: RRID:CVCL_1G42
RRID:CVCL_B478
DOI: 10.1371/journal.pone.0348959
Resource: (NIH-ARP Cat# 8129-442, RRID:CVCL_B478)
Curator: @scibot
SciCrunch record: RRID:CVCL_B478
RRID:AB_314519
DOI: 10.1371/journal.pone.0348959
Resource: (BioLegend Cat# 305311, RRID:AB_314519)
Curator: @scibot
SciCrunch record: RRID:AB_314519
RRID:AB_2890803
DOI: 10.1371/journal.pone.0348959
Resource: (BioLegend Cat# 370611, RRID:AB_2890803)
Curator: @scibot
SciCrunch record: RRID:AB_2890803
RRID:AB_2721574
DOI: 10.1371/journal.pone.0348959
Resource: (BioLegend Cat# 374204, RRID:AB_2721574)
Curator: @scibot
SciCrunch record: RRID:AB_2721574
RRID:CVCL_0063
DOI: 10.1371/journal.pone.0348959
Resource: (RRID:CVCL_0063)
Curator: @scibot
SciCrunch record: RRID:CVCL_0063
BDSC_80435
DOI: 10.1371/journal.pbio.3002843
Resource: RRID:BDSC_80435
Curator: @scibot
SciCrunch record: RRID:BDSC_80435
BDSC_51794
DOI: 10.1371/journal.pbio.3002843
Resource: RRID:BDSC_51794
Curator: @scibot
SciCrunch record: RRID:BDSC_51794
Plasmid_139447
DOI: 10.1210/endocr/bqag073
Resource: RRID:Addgene_139447
Curator: @scibot
SciCrunch record: RRID:Addgene_139447
Plasmid_23252
DOI: 10.1210/endocr/bqag073
Resource: RRID:Addgene_23252
Curator: @scibot
SciCrunch record: RRID:Addgene_23252
RRID:AB_2231996
DOI: 10.1210/endocr/bqag073
Resource: (Santa Cruz Biotechnology Cat# sc-253, RRID:AB_2231996)
Curator: @scibot
SciCrunch record: RRID:AB_2231996
RRID:AB_2097174
DOI: 10.1210/endocr/bqag073
Resource: (Santa Cruz Biotechnology Cat# sc-110, RRID:AB_2097174)
Curator: @scibot
SciCrunch record: RRID:AB_2097174
RRID:SCR_001905
DOI: 10.1200/PO-25-01229
Resource: R Project for Statistical Computing (RRID:SCR_001905)
Curator: @scibot
SciCrunch record: RRID:SCR_001905
RRID:SCR_020239
DOI: 10.1200/PO-25-01229
Resource: Life Technologies QuantStudio 6 Real Time PCR System (RRID:SCR_020239)
Curator: @scibot
SciCrunch record: RRID:SCR_020239
RRID:AB_477010
DOI: 10.1186/s40478-026-02240-y
Resource: (Sigma-Aldrich Cat# G3893, RRID:AB_477010)
Curator: @scibot
SciCrunch record: RRID:AB_477010
RRID:AB_2572311
DOI: 10.1186/s40478-026-02240-y
Resource: (EnCor Biotechnology Cat# MCA-5C10, RRID:AB_2572311)
Curator: @scibot
SciCrunch record: RRID:AB_2572311
RRID:AB_3678889
DOI: 10.1186/s40478-026-02240-y
Resource: RRID:AB_3678889
Curator: @scibot
SciCrunch record: RRID:AB_3678889
RRID:AB_2109953
DOI: 10.1186/s40478-026-02240-y
Resource: (EnCor Biotechnology Cat# CPCA-GFAP, RRID:AB_2109953)
Curator: @scibot
SciCrunch record: RRID:AB_2109953
RRID:AB_2050678
DOI: 10.1186/s40478-026-02240-y
Resource: (Enzo Life Sciences Cat# BML-FG6090-0100, RRID:AB_2050678)
Curator: @scibot
SciCrunch record: RRID:AB_2050678
RRID:AB_3678888
DOI: 10.1186/s40478-026-02240-y
Resource: RRID:AB_3678888
Curator: @scibot
SciCrunch record: RRID:AB_3678888
RRID:AB_3711244
DOI: 10.1186/s40478-026-02240-y
Resource: RRID:AB_3711244
Curator: @scibot
SciCrunch record: RRID:AB_3711244
RRID:AB_10013382
DOI: 10.1186/s40478-026-02240-y
Resource: (Agilent Cat# Z0334, RRID:AB_10013382)
Curator: @scibot
SciCrunch record: RRID:AB_10013382
RRID:SCR_003070
DOI: 10.1186/s13058-026-02307-7
Resource: ImageJ (RRID:SCR_003070)
Curator: @scibot
SciCrunch record: RRID:SCR_003070
RRID:CVCL_0493
DOI: 10.1186/s13058-026-02307-7
Resource: (ATCC Cat# TIB-71, RRID:CVCL_0493)
Curator: @scibot
SciCrunch record: RRID:CVCL_0493
RRID:CVCL_J239
DOI: 10.1186/s13058-026-02307-7
Resource: (JCRB Cat# JCRB1447, RRID:CVCL_J239)
Curator: @scibot
SciCrunch record: RRID:CVCL_J239
RRID:CVCL_0062
DOI: 10.1186/s13058-026-02307-7
Resource: (RRID:CVCL_0062)
Curator: @scibot
SciCrunch record: RRID:CVCL_0062
RRID:SCR_002798
DOI: 10.1186/s13058-026-02307-7
Resource: GraphPad Prism (RRID:SCR_002798)
Curator: @scibot
SciCrunch record: RRID:SCR_002798
RRID:SCR_001905
DOI: 10.1186/s12957-026-04511-3
Resource: R Project for Statistical Computing (RRID:SCR_001905)
Curator: @scibot
SciCrunch record: RRID:SCR_001905
RRID:SCR_006902
DOI: 10.1186/s12957-026-04511-3
Resource: Surveillance Epidemiology and End Results (RRID:SCR_006902)
Curator: @scibot
SciCrunch record: RRID:SCR_006902
RRID:AB_2099233
DOI: 10.1177/00368504261470488
Resource: (Cell Signaling Technology Cat# 7074, RRID:AB_2099233)
Curator: @scibot
SciCrunch record: RRID:AB_2099233
RRID:AB_2223172
DOI: 10.1177/00368504261470488
Resource: (Cell Signaling Technology Cat# 4970, RRID:AB_2223172)
Curator: @scibot
SciCrunch record: RRID:AB_2223172
RRID:AB_2491009
DOI: 10.1177/00368504261470488
Resource: (Cell Signaling Technology Cat# 9145, RRID:AB_2491009)
Curator: @scibot
SciCrunch record: RRID:AB_2491009
RRID:SCR_008567
DOI: 10.1158/1940-6207.CAPR-26-0044
Resource: Statistical Analysis System (RRID:SCR_008567)
Curator: @scibot
SciCrunch record: RRID:SCR_008567
RRID:SCR_012763
DOI: 10.1158/1940-6207.CAPR-26-0044
Resource: Stata (RRID:SCR_012763)
Curator: @scibot
SciCrunch record: RRID:SCR_012763
RRID:SCR_012429
DOI: 10.1158/1940-6207.CAPR-26-0044
Resource: Arizona University Genetics Core Facility (RRID:SCR_012429)
Curator: @scibot
SciCrunch record: RRID:SCR_012429
RRID:SCR_003445
DOI: 10.1158/1940-6207.CAPR-26-0044
Resource: REDCap (RRID:SCR_003445)
Curator: @scibot
SciCrunch record: RRID:SCR_003445
SCR_006659
DOI: 10.1158/1940-6207.CAPR-26-0044
Resource: University of Pittsburgh; Pennsylvania; USA (RRID:SCR_006659)
Curator: @scibot
SciCrunch record: RRID:SCR_006659
RRID:SCR_010605
DOI: 10.1158/1940-6207.CAPR-26-0044
Resource: RRID:SCR_010605
Curator: @scibot
SciCrunch record: RRID:SCR_010605
RRID:SCR_000339
DOI: 10.1158/1940-6207.CAPR-26-0044
Resource: RRID:SCR_000339
Curator: @scibot
SciCrunch record: RRID:SCR_000339
RRID:SCR_012866
DOI: 10.1158/1940-6207.CAPR-26-0044
Resource: RRID:SCR_012866
Curator: @scibot
SciCrunch record: RRID:SCR_012866
RRID:SCR_001905
DOI: 10.1158/1055-9965.EPI-25-2008
Resource: R Project for Statistical Computing (RRID:SCR_001905)
Curator: @scibot
SciCrunch record: RRID:SCR_001905
RRID:Addgene_85168
DOI: 10.1128/aem.02536-25
Resource: RRID:Addgene_85168
Curator: @scibot
SciCrunch record: RRID:Addgene_85168
Addgene_12260
DOI: 10.1093/nar/gkag728
Resource: RRID:Addgene_12260
Curator: @scibot
SciCrunch record: RRID:Addgene_12260
plasmid_216279
DOI: 10.1093/nar/gkag728
Resource: RRID:Addgene_216279
Curator: @scibot
SciCrunch record: RRID:Addgene_216279
plasmid_200639
DOI: 10.1093/nar/gkag728
Resource: RRID:Addgene_200639
Curator: @scibot
SciCrunch record: RRID:Addgene_200639
plasmid_12259
DOI: 10.1093/nar/gkag728
Resource: RRID:Addgene_12259
Curator: @scibot
SciCrunch record: RRID:Addgene_12259
RRID:SCR_016477
DOI: 10.1093/cercor/bhag071
Resource: CERVO Canadian Optogenetics and Vectorology Foundry Core Facility (RRID:SCR_016477)
Curator: @scibot
SciCrunch record: RRID:SCR_016477
SAMN15879419
DOI: 10.1080/19382014.2026.2706228
Resource: RRID:SAMN15879419
Curator: @scibot
SciCrunch record: RRID:SAMN15879419
SAMN31759877
DOI: 10.1080/19382014.2026.2706228
Resource: RRID:SAMN31759877
Curator: @scibot
SciCrunch record: RRID:SAMN31759877
SAMN15879346
DOI: 10.1080/19382014.2026.2706228
Resource: RRID:SAMN15879346
Curator: @scibot
SciCrunch record: RRID:SAMN15879346
SAMN15879404
DOI: 10.1080/19382014.2026.2706228
Resource: RRID:SAMN15879404
Curator: @scibot
SciCrunch record: RRID:SAMN15879404
RRID:AB_2340621
DOI: 10.1080/19382014.2026.2706228
Resource: (Jackson ImmunoResearch Labs Cat# 711-585-152, RRID:AB_2340621)
Curator: @scibot
SciCrunch record: RRID:AB_2340621
SAMN39996136
DOI: 10.1080/19382014.2026.2706228
Resource: RRID:SAMN39996136
Curator: @scibot
SciCrunch record: RRID:SAMN39996136
RRID:AB_2340854
DOI: 10.1080/19382014.2026.2706228
Resource: (Jackson ImmunoResearch Labs Cat# 715-585-150, RRID:AB_2340854)
Curator: @scibot
SciCrunch record: RRID:AB_2340854
SAMN15879290
DOI: 10.1080/19382014.2026.2706228
Resource: RRID:SAMN15879290
Curator: @scibot
SciCrunch record: RRID:SAMN15879290
RRID:AB_2538613
DOI: 10.1080/19382014.2026.2706228
Resource: (Thermo Fisher Scientific Cat# MA5-17142, RRID:AB_2538613)
Curator: @scibot
SciCrunch record: RRID:AB_2538613
RRID:AB_881819
DOI: 10.1080/19382014.2026.2706228
Resource: (Abcam Cat# ab54210, RRID:AB_881819)
Curator: @scibot
SciCrunch record: RRID:AB_881819
RRID:AB_2800361
DOI: 10.1080/19382014.2026.2706228
Resource: (Agilent Cat# IR002, RRID:AB_2800361)
Curator: @scibot
SciCrunch record: RRID:AB_2800361
RRID:SCR_014641
DOI: 10.1080/19382014.2026.2706228
Resource: Network for Pancreatic Organ Donors with Diabetes (RRID:SCR_014641)
Curator: @scibot
SciCrunch record: RRID:SCR_014641
RRID:AB_2340472
DOI: 10.1080/19382014.2026.2706228
Resource: (Jackson ImmunoResearch Labs Cat# 706-545-148, RRID:AB_2340472)
Curator: @scibot
SciCrunch record: RRID:AB_2340472
RRID:AB_628067
DOI: 10.1080/19382014.2026.2706228
Resource: (Santa Cruz Biotechnology Cat# sc-1661, RRID:AB_628067)
Curator: @scibot
SciCrunch record: RRID:AB_628067
RRID:SCR_019170
DOI: 10.1073/pnas.2603906123
Resource: University of North Carolina at Chapel Hill Flow Cytometry Core Facility (RRID:SCR_019170)
Curator: @scibot
SciCrunch record: RRID:SCR_019170
RRID:SCR_022168
DOI: 10.1073/pnas.2603906123
Resource: North Carolina State University High Performance Computing Services Core Facility (RRID:SCR_022168)
Curator: @scibot
SciCrunch record: RRID:SCR_022168
RRID:Addgene_66399
DOI: 10.1038/s44386-026-00058-1
Resource: RRID:Addgene_66399
Curator: @scibot
SciCrunch record: RRID:Addgene_66399
